
Overview of sequencing and De Novo assembly
We analysed 36 composite samples from two cultivars with contrasting yields to profile the ginger-associated microbiomes. High-throughput sequencing generated 1.32 million quality-filtered reads for bacteria/archaea and 1.22 million for fungi, identifying 5970 bacterial/archaeal OTUs and 1701 fungal OTUs. Bacterial communities spanned 46 phyla, while fungal taxa were classified into 17 phyla, as annotated against the SILVA and UNITE databases (Supplementary Data 1). Rarefaction curves (Supplementary Fig. 1) and Shannon diversity indices (Supplementary Fig. 2) confirmed that sequencing depth was sufficient to capture most community diversity. Bacterial richness was more than threefold higher than fungi’s, highlighting their broader taxonomic diversity across soil and plant compartments.
Contrasting assembly mechanisms in bacterial and fungal communities
We applied the neutral community model to elucidate the mechanisms driving microbial community assembly in two ginger varieties with distinct yield characteristics. Our analysis revealed contrasting assembly patterns between bacterial and fungal communities. Bacterial communities were predominantly shaped by stochastic processes, as evidenced by strong fits to the neutral model (R² = 0.67 and 0.68) and high migration rates (Nm = 1353 and 1440) for both cultivars (Fig. 1a). In contrast, fungal communities exhibited poor fits to the neutral model (R² = −0.03 and −0.08), indicating a more substantial influence of deterministic processes (Fig. 1b). To dissect these assembly mechanisms further, we calculated the β-nearest-taxon index (βNTI), which assesses niche-level assembly processes. Bacterial communities displayed elevated mean βNTI values in both cultivars’ rhizosphere and root tissues, pointing to a dominant deterministic influence in these niches (Fig. 1c). Conversely, fungal communities showed evidence of deterministic assembly, with strong homogeneous selection (βNTI < -2) across various plant tissues (Fig. 1d).
Fig. 1: Ecological processes governing the assembly of bacterial and fungal communities across microbial niches in two ginger cultivars with contrasting yields.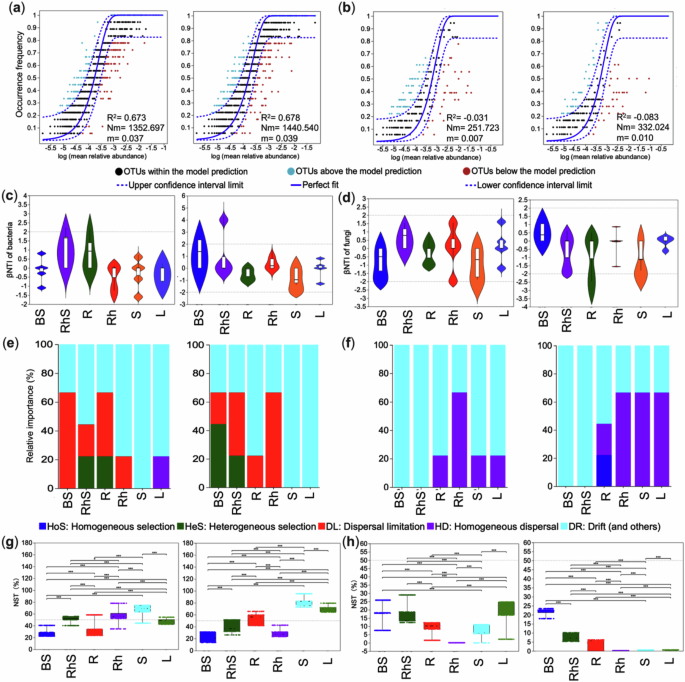
This figure illustrates the relative influence of stochastic versus deterministic forces on community assembly for bacterial and fungal microbiomes. Neutral community model (NCM) fits for bacterial (a) and fungal (b) communities. The solid blue line indicates the Sloan model fit, with dashed lines denoting the 95% confidence intervals. Operational taxonomic units (OTUs) falling significantly above or below the model are shown in green and red, respectively. Model performance is summarised by the coefficient of determination (R²) and the product of metacommunity size and immigration rate (Nm). Violin plots of the β-nearest taxon index (βNTI) for bacterial (c) and fungal (d) communities. Values of |βNTI|≥ 2 indicate deterministic assembly, while |βNTI|< 2 suggests stochasticity. Relative contributions of distinct ecological processes to bacterial (e) and fungal (f) community assembly, inferred from βNTI and Bray–Curtis–based Raup–Crick index (RCBray).Normalised stochasticity ratio (NST) for bacterial (g) and fungal (h) communities across compartments. The NST threshold of 0.5 (dashed line) separates predominantly deterministic (<0.5) from stochastic (>0.5) dynamics. BS bulk soil, RhS rhizosphere soil, R root, Rh rhizome, S stem, L leaf. Panels are split by cultivar: variety one (left) and variety two (right).
Compositional analyses revealed significant niche-specific variations in bacterial communities (Fig. 1e). In the first variety’s bulk soil, community structure was mainly shaped by dispersal limitation (66.67%). In contrast, in the second variety, heterogeneous selection was more dominant (44.44%). Both varieties exhibited substantial drift effects in stem and leaf samples, highlighting the diversity of assembly processes across niches. Fungal community assembly, however, followed a different trajectory. While stochastic processes dominated in bulk and rhizosphere soils (100%), root tissues displayed a mix of influences. The roots of the first variety were primarily shaped by drift (77.78%), whereas those of the second variety were influenced by a combination of drift, homogeneous dispersal, and selection (Fig. 1f).
We used the normalised stochasticity ratio (NST) to evaluate the balance between stochastic and deterministic assembly, with 50% as the threshold. Bacterial endophytic communities exhibited considerable stochasticity (NSTavg > 0.5), particularly in the rhizomes and stems of the first variety (Fig. 1g). In contrast, fungal communities in both cultivars were predominantly governed by deterministic processes (Fig. 1h).
Distinct ecological processes shaped bacterial communities in different plant compartments. Deterministic selection dominated the rhizosphere and endosphere, while stochastic influences were more evident in the bulk soil. Fungal communities in roots shifted from drift in the low-yield variety to mixed deterministic processes in the high-yield counterpart.
Host genotype modulates niche-specific microbial structuring
Because the two varieties were grown in different locations, standardised cultivation practices and synchronised sampling were applied to reduce potential environmental variation. This design allows observed microbiome and metabolome differences to be more confidently attributed to host genotype rather than site-specific management effects.
A comprehensive analysis revealed that microbial communities in ginger are strongly compartmentalised and influenced by host genotype. Kruskal‒Wallis test indicated a significant reduction in alpha diversity (P < 0.05) in aerial tissues relative to soil and subterranean compartments across both varieties (Fig. 2a, b), underscoring the importance of the root-soil interface in sustaining microbial richness—likely due to its greater nutrient heterogeneity and physical complexity. Principal coordinate analysis (PCoA) based on Bray‒Curtis dissimilarity confirmed clear niche segregation in bacterial communities, which was statistically supported by Adonis testing (R² = 0.7241 for variety one; R² = 0.6908 for variety two; P = 0.001) (Fig. 2c).
Fig. 2: Alpha and beta diversity of bacterial and fungal communities across microbial niches in two ginger varieties differing in yield.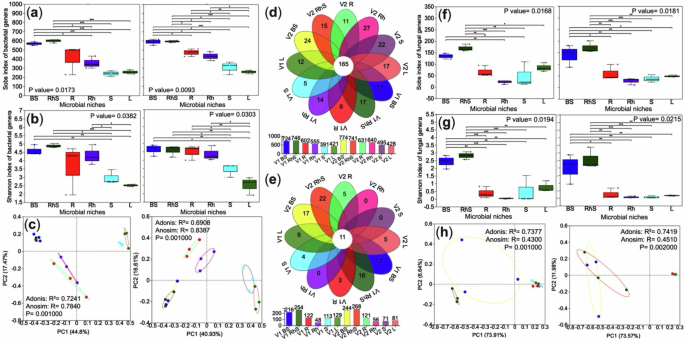
This figure summarises microbial diversity patterns and niche-specific taxa in bulk soil, rhizosphere, and internal tissues. a, b Alpha diversity of bacterial communities based on the observed species richness (Sobs) and Shannon index. c Principal coordinate analysis (PCoA) of bacterial beta diversity using Bray–Curtis distances, with statistical significance assessed by Adonis/PERMANOVA. Venn diagrams showing shared (central ring) and unique bacterial (d) and fungal (e) genera across all compartments, with bar charts indicating the total number of genera per niche. Details on niche-specific genera are provided in Supplementary Data. Fungal alpha diversity was evaluated using Sobs (f) and Shannon (g) indices. h PCoA of fungal beta diversity with significance tested by Adonis/PERMANOVA. All comparisons are presented in the figures, and comprehensive statistical results are available in Supplementary Data. Statistical differences were assessed using the Kruskal–Wallis test with Benjamini–Hochberg correction; significance is denoted as p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***). Sample type BS bulk soil, RhS rhizosphere soil, R root, Rh rhizome, S stem, L leaf. Panels are grouped by cultivar: variety one (left) and variety two (right).
Notably, the endophytic communities associated with the underground environment of variety two exhibited more significant dissimilarity than those of variety one (Fig. 2c), suggesting varietal-specific influences on microbial assembly. Microbial composition analysis, visualised through floral and bar graphs, identified generalist and niche-specific microbial taxa. The high-yield variety hosted 774 bacterial genera in bulk soil (vs. 746 in low-yield) and enriched nitrogen-fixing Devosia (see Fig. 2d–e and Supplementary Data 2). Fungal communities were niche-specialised, with only 0.64% generalists (Fig. 2f–h). Dominant fungal genera were highly adapted to the unique soil microbiomes of each variety, suggesting limited potential for direct colonisation of host tissues. This finding underscores the role of the root system as a selective interface, filtering and shaping microbial communities. The distinct separation between soilborne and endophytic communities (Fig. 2f, g) was further supported by unique beta diversity patterns (Fig. 2h), emphasising the strong influence of the root system on microbial assembly processes.
Host metabolites shape microbiome composition and function
To investigate how host metabolites influence microbiome assembly and yield outcomes, we performed untargeted LC–MS/MS profiling across ginger tissues from two cultivars. A total of 586 unique metabolites were identified, with 511 matched to spectral libraries and 200 annotated in KEGG pathways. Principal component analysis (PCA) revealed clear separation of metabolite profiles according to both plant tissue type and cultivar yield level (Fig. 3a). Samples from the high-yield cultivar clustered distinctly from the low-yield cultivar along PC1, which explained 57.1% of the total variance, indicating cultivar-specific metabolic reprogramming. PC2 (27.4% variance) primarily separated tissues, with rhizome and root samples forming tighter clusters relative to leaf and stem samples. These results suggest that genotype and tissue identity shape the ginger metabolome, with yield-associated varieties exhibiting coordinated shifts in primary and secondary metabolite production.
Fig. 3: Host metabolite profiles and microbe–metabolite associations in ginger cultivars with contrasting yields.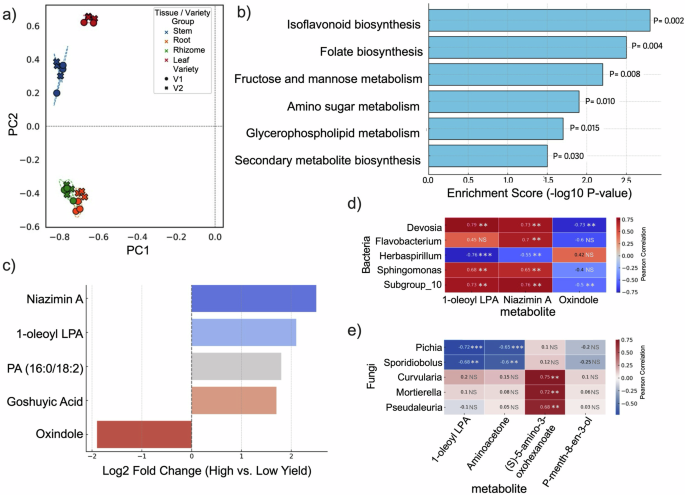
a Principal component analysis (PCA) of untargeted LC-MS/MS metabolite profiles, showing separation by tissue type and cultivar (V1: low yield; V2: high yield). b KEGG pathway enrichment in high-yield tissues reveals significant upregulation of isoflavonoid and folate biosynthesis, among other metabolic pathways. c Differentially abundant metabolites between high- and low-yield cultivars, including Niazimin A, 1-oleoyl lysophosphatidic acid (LPA), and Oxindole. d The Pearson correlation heatmap shows strong positive associations between host metabolites and growth-promoting bacteria (e.g., Devosia) and negative associations with Oxindole. e The correlation heatmap of key host metabolites and fungal taxa indicates differential microbial responses and potential host-driven filtering. Numbers in each cell indicate Pearson correlation coefficients. Statistical significance is indicated by asterisks (*p < 0.05, **p < 0.01, ***p < 0.001).
KEGG pathway enrichment analysis revealed that several biosynthetic and metabolic pathways were significantly upregulated in the high-yield ginger cultivar (Fig. 3b). Notably, isoflavonoid biosynthesis and folate metabolism were among the most enriched, both of which are implicated in plant defence and growth. Additional pathways included fructose and mannose metabolism, amino sugar metabolism, and glycerophospholipid metabolism, suggesting broad-scale carbon and nitrogen flow reprogramming. Enriching secondary metabolite biosynthesis pathways aligns with the observed increase in yield-associated microbial taxa, supporting a potential role for plant-derived compounds in shaping beneficial microbiome composition.
Differential metabolite analysis identified several compounds with significantly higher abundance in the high-yield cultivar (VIP > 1, FDR < 0.05; Fig. 3c). Among these, Niazimin A (an alkaloid), 1-oleoyl lysophosphatidic acid (a lipid mediator), and PA (16:0/18:2) were particularly enriched in rhizome and root tissues. Goshuyic acid also showed consistent upregulation across tissues. In contrast, oxindole was more abundant in the low-yield cultivar and may be linked to microbial or metabolic signatures of suboptimal performance. The identity and distribution of these metabolites suggest that they serve as biochemical signals, influencing the microbiome assembly, nutrient exchange, or host defence. To evaluate the ecological roles of these compounds, we analysed the correlations between metabolite concentrations and microbial abundances in various tissues. In the high-yielding cultivar, 1-oleoyl lysophosphatidic acid was strongly associated with Devosia and Subgroup_10 (r > 0.75, P < 0.01), which are involved in nitrogen fixation. Niazimin A correlated with Sphingomonas, while (S)-5-amino-3-oxohexanoate was positively associated with Flavobacterium and Devosia. Conversely, the metabolite oxindole—more abundant in the low-yield cultivar—showed negative correlations with these same taxa (Fig. 3d).
Fungal community structure also mirrored these metabolite patterns. In the low-yield cultivar, (S)-5-amino-3-oxohexanoate positively correlated with Curvularia, Mortierella, and Pseudaleuria, while Apiotrichum showed negative associations with P-menth-8-en-3-ol. In the high-yielding cultivar, Pichia and Sporidiobolus were negatively associated with 1-oleoyl lysophosphatidic acid and aminoacetone (Fig. 3e).
These findings indicate that metabolites produced by the host serve as selective filters, influencing the diversity and activity of bacterial and fungal taxa associated with nutrient cycling, disease suppression, and the plant’s overall performance.
Keystone Taxa and network complexity underlie yield differences
We focused on dominant operational taxonomic units (OTUs) with a relative abundance of ≥2% to identify keystone microbial taxa within the ginger microbiome. We pinpointed potential biomarker taxa using linear discriminant analysis effect size (LEfSe) and differential abundance analyses. Network analysis further revealed keystone taxa based on their topological roles:
Bacterial nodes were classified as network hubs if they exhibited a degree >13 and a closeness centrality >0.08, while fungal nodes required a degree >42 and a closeness centrality >0.22. These criteria identified taxa with central roles in microbial networks, suggesting their potential as keystone representatives within the ginger microbiome.
In this study, hub and keystone taxa are related but not synonymous. We define hub taxa based on network topology — specifically, nodes with high degree and closeness centrality values, indicating structural centrality within co-occurrence networks. In contrast, keystone taxa refer to microbial taxa with a disproportionately large impact on community structure or function, as supported by multi-omics correlations, ecological relevance, or enrichment in high-yield systems. While some taxa may fulfil both criteria, we use these terms with context-specific precision to distinguish between structural centrality and ecological function.
Taxonomic and functional differentiation of ginger-associated microbial communities
Comparative analysis of microbial communities in two ginger varieties revealed striking taxonomic and functional divergences that correlate with yield variation and niche adaptation.
Genus-level resolution identified Sphingomonas, Methylobacterium-Methylorubrum, and Bacillus as core taxa across both varieties. Notably, Flavobacterium was enriched in variety one (4.73%), whereas Acidovorax was more prevalent in variety two (3.14%). Tissue-specific patterns highlighted clear microbial compartmentalisation: Sphingomonas dominated leaf tissues, while Bacillus was most abundant in bulk soil. The nitrogen-fixing Allorhizobium-Neorhizobium-Pararhizobium-Rhizobium complex exhibited niche preference for stems in variety one and roots/rhizomes in variety two. Remarkably, Flavobacterium reached 22.91% abundance in the roots of variety one (Fig. 4a).
Fig. 4: Composition, biomarkers, and functional profiles of bacterial and fungal communities across ginger varieties and compartments.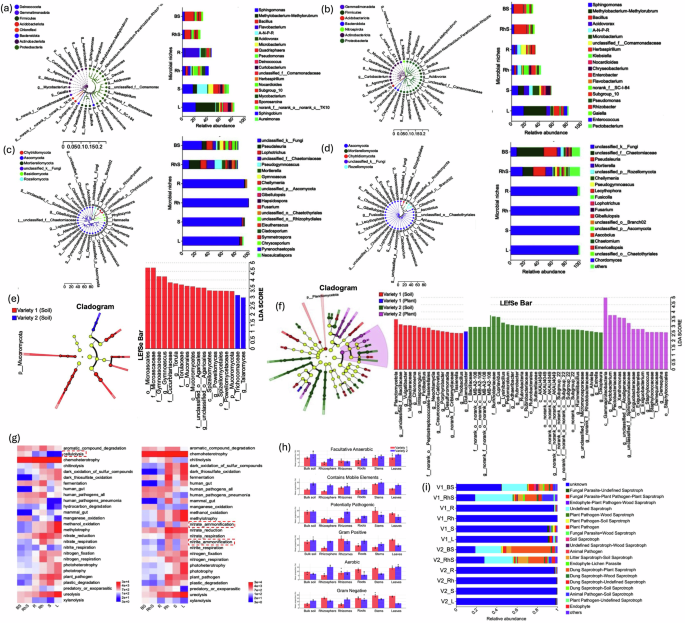
This multi-panel figure integrates taxonomic composition, indicator taxa, and predicted ecological functions of bacterial and fungal communities associated with two ginger varieties. a, b Relative abundance of the top 20 bacterial genera across tissues and varieties, visualized as stacked bar plots. c, d The relative abundance of the top 20 fungal genera is structured similarly (a, c V1: low yield; b, d, V2: high yield). Discriminatory taxa identified by LEfSe analysis (LDA > 2.5, P < 0.05), showing bacterial (e) and fungal (f) biomarkers between varieties, displayed as LDA score plots and cladograms. g Functional prediction of bacterial communities based on FAPROTAX, highlighting nitrogen-related processes such as nitrate/nitrite ammonification and nitrate reduction. h BugBase-derived prediction of bacterial functional groups. Bars show mean ± SD. Individual points represent biologically independent replicates (n = 3). i Functional guild classification of fungal communities via FUNGuild, distinguishing dominant trophic strategies across compartments. BS bulk soil, RhS rhizosphere soil, R root, Rh rhizome, S stem, L leaf, V1 variety one, V2 variety two.
Fungal communities were primarily composed of unclassified taxa (>68% in both varieties), with Ascomycota as the dominant phylum. Pseudaleuria and Lophotrichus emerged as prevalent genera (Fig. 4b). LEfSe analysis further identified variety-specific fungal biomarkers such as Gymnoascus, Torula, and Powellomyces for variety one and Talaromyces for variety two, highlighting divergent evolutionary trajectories in rhizosphere adaptation (Fig. 4c). Microbial biomarker analysis revealed 24 discriminatory bacterial genera (LDA > 2.5, P < 0.05). Variety one was characterised by soilborne taxa such as Vulgatibacter and Intrasporangium. In contrast, variety two was enriched in Cupriavidus, Pectobacterium, and Rubrobacter (Fig. 4d). Despite these taxonomic differences, functional predictions using FAPROTAX suggested only modest divergence. Variety one showed higher cellulolytic activity, while variety two displayed enriched nitrogen metabolism, particularly in rhizome and foliar compartments, including ammonification and nitrate reduction pathways (Fig. 4e).
Phenotypic predictions using BugBase distinguished functional traits between niches. Variety two exhibited traits associated with facultative anaerobes, especially in rhizomes and stems (Fig. 4f). FUNGuild analysis corroborated these distinctions, revealing differential representation of saprotrophs and pathogens between tissue types and varieties (Fig. 4g).
These findings highlight the significant influence of host genotype and environmental conditions on the composition and functional capabilities of microbial communities associated with ginger. By integrating taxonomic identification and metabolic analysis, we better understand how microbiomes contribute to yield and resilience in ginger cultivation systems.
Microbial cooccurrence networks and the identification of keystone microbial Taxa
Network analysis of the 50 most abundant microbial species revealed significant structural differences (Pearson r > 0.96; P < 0.05) between the two ginger varieties. In variety one, the bacterial microbiome was characterized by 163 nodes and 272 edges (see Fig. 5a). In comparison, variety two displayed a more complex network consisting of 155 nodes and 596 edges (see Fig. 5b). Notably, there were no negative interactions among the microbial taxa in either ecosystem, highlighting a predominant trend of symbiotic relationships within these communities.
Fig. 5: Microbial co-occurrence networks and keystone taxa in ginger varieties with contrasting yields.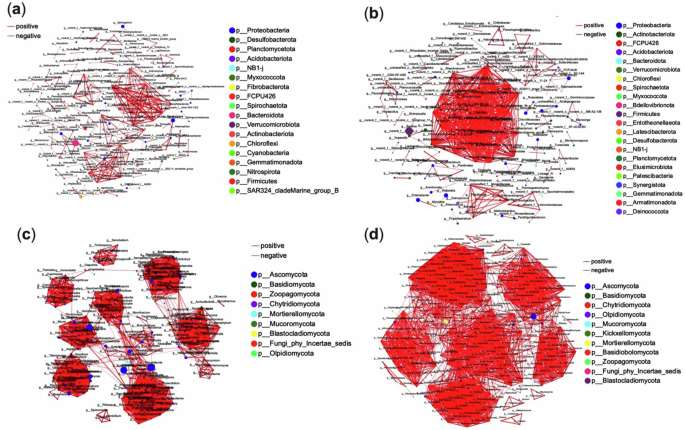
Co-occurrence networks illustrate the structural organisation of dominant bacterial (a, b) and fungal (c, d) genera in low-yield (a, c) and high-yield (b, d) ginger cultivars. Nodes represent genera, colour-coded by phylum, with node size proportional to degree (number of connections). Edges represent significant pairwise correlations (Pearson’s r > 0.96, P < 0.05): red for positive, green for negative. Keystone taxa (network hubs) were defined by high degree centrality, indicating potential roles in stabilising or organising microbial community structure.
Variety two demonstrated greater complexity in bacterial interactions, with an edge-to-node ratio of 3.84 compared to 1.67 in variety one. The mean connectivity of variety two (7.65) also exceeded that of variety one (3.34), suggesting the presence of highly interconnected modules within its network. Furthermore, centralisation and density metrics were higher in variety two, reflecting a more connected and organised microbial structure. Eleven network hubs, including Ulvibacter, Saccharothrix, and Paenalcaligenes, were identified in variety one. In contrast, variety two harboured 27 network hubs, such as Dialister, Lachnospira, and Fusicatenibacter. These hub taxa represent keystone species critical for maintaining ecological integrity and functional stability.
Fungal co-occurrence networks also revealed greater complexity in variety two, with an edge-to-node ratio of 12.23 (Fig. 5d) compared to 11.04 in variety one (Fig. 5c). Keystone fungal taxa differed significantly between the varieties: variety one was characterised by Myrmecridium, Pseudorobillarda, Duddingtonia, and Chalara, while variety two featured Microascus, Ganoderma, Talaromyces, and Leucothecium, among others.
These findings underscore microbial communities’ intricate and diverse nature, which is shaped by the ecological contexts of different ginger varieties. The discrepancies in keystone taxa highlight their potential influence on ecosystem dynamics, including nutrient interactions and microbial co-occurrence patterns. Further analyses will explore the functional roles of these keystone taxa and their contributions to the stability and resilience of ginger-associated microbial networks. Network analysis revealed the high-yield variety’s microbiome was more interconnected, with 27 bacterial hubs (e.g., Dialister, Lachnospira) versus 11 in low-yield. Fungal hubs (Talaromyces, Ganoderma) in the high-yield variety correlated with nutrient turnover.
Nutrient dynamics mediate microbial recruitment
The relationships between nutrient dynamics and microbial community co-occurrences in ginger were investigated by analysing physicochemical parameters of soil and plant samples (see Supplementary Table 1). Multicollinearity was assessed using the variance inflation factor (VIF) criterion, excluding factors with VIF > 10. In the first ginger variety, four parameters – total nitrogen (TN), total potassium (TK), total phosphorus (TP), and available potassium (AK) – were correlated with microbial communities. In contrast, the second variety included five parameters: TN, TK, TP, AK, and available phosphorus (AP). Distance-based redundancy analysis (db-RDA) was used to evaluate the influence of these nutrient factors on community structure. These variables explained approximately 20% of the variation in the assemblages of the 25 most prevalent bacterial (Fig. 6a) and fungal (Fig. 6b) genera. Significant genus-level associations were identified between nutrient parameters and distinct microbiomes across both varieties.
Fig. 6: Nutrient–microbiome interactions across ginger cultivars with contrasting yields.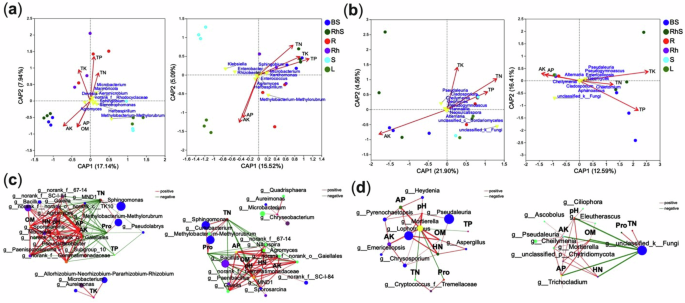
Distance-based redundancy analysis (db-RDA) showing the influence of nutrient concentrations on bacterial (a) and fungal (b) community composition. Coloured points represent microbial niches, red arrows indicate nutrient vectors, and yellow labels denote responsive microbial taxa. Microbial–nutrient correlation networks for bacterial (c) and fungal (d) taxa. Nodes represent microbial genera, with size proportional to relative abundance and colour-coded by taxonomy. Edges represent significant correlations (r > 0.7, P < 0.05): red for positive, green for negative. Edge thickness reflects correlation strength. BS bulk soil, RhS rhizosphere soil, R root, Rh rhizome, S stem, L leaf. Variety one (left panels), variety two (right panels).
In the first variety, AK strongly correlated with the soil bacterial microbiome, TK with the rhizome microbiome, and TP and TN with the root microbiome (Fig. 6a, P < 0.05). AK and AP were associated with the leaf microbiome in the second variety, while TP, TN, and TK correlated with the soil microbiome (Fig. 6a, P < 0.05).
For fungal communities, AK correlated with the soil microbiome, and TP with the root and stem microbiomes in variety one (Fig. 6b, P < 0.05). In variety two, AK and AP were linked to the root, rhizome, stem, and leaf microbiomes, while TK and TN were associated with the rhizosphere microbiome, and TP with the bulk soil microbiome (Fig. 6b, P < 0.05). Co-occurrence networks revealed significant correlations (Pearson’s r ≥ 0.85, P < 0.05) between nutrient parameters and microbial taxa. The bacterial network comprised 38 nodes for both varieties, while the fungal network included 13 nodes for variety one and 19 for variety two.
In variety one, 27 bacterial taxa showed negative correlations with TN, while two were positively correlated. AK was positively associated with 22 taxa, TK with three, and TP negatively with 14. In variety two, TN negatively correlated with 20 genera and positively with three, while AK, AP, and TK positively associated with 15, 17, and five genera, respectively.
Key taxa were identified through node degree and closeness centrality analyses. For example, Solirubrobacter (r = 0.94), Paenisporosarcina (r = 0.93), and Agromyces (r = 0.93) positively correlated with AK (P < 0.01). Microbacterium (r = 0.84) and Aureimonas (r = 0.76) were positively associated with TK, while Bacillus (r = −0.71), Nitrospira (r = −0.74), and Agromyces (r = -0.76) negatively correlated with TN (P < 0.01). Cryobacterium (r = -0.70), Solirubrobacter (r = −0.71), and Microvirga (r = −0.72) were negatively correlated with TP (P < 0.01).
In variety two, Agromyces (r = 0.88), Nitrospira (r = 0.84), and Bryobacter (r = 0.82) positively correlated with AK (P < 0.01). Quadrisphaera (r = 0.81), Klenkia (r = 0.76), Chryseobacterium (r = 0.73), and Aureimonas (r = 0.73) were positively associated with TK (P < 0.01). Curtobacterium (r = 0.81) and Methylobacterium-Methylorubrum (r = 0.75) positively correlated with TN, while Reyranella (r = −0.72), Agromyces (r = −0.76), Nitrospira (r = −0.77), and Solirubrobacter (r = −0.79) showed negative correlations (P < 0.01). Bryobacter (r = −0.71) was negatively correlated with TP, while MND1 (r = 0.92), Nitrospira (r = 0.91), Gaiella (r = 0.89), Solirubrobacter (r = 0.84), and Agromyces (r = 0.84) positively correlated with AP (P < 0.01) (Fig. 6d).
For fungal communities in variety one, Mortierella (r = 0.83), Chrysosporium (r = 0.79), Pyrenochaetopsis (r = 0.77), Emericellopsis (r = 0.75), and Lophotrichus (r = 0.70) positively correlated with AK (P < 0.01). Mortierella was negatively correlated with TN (r = −0.78) and TP (r = −0.72) (P < 0.01). In variety two, Chrysosporium (r = 0.91), Penicillium (r = 0.81), Cheilymenia (r = 0.79), and Podospora (r = 0.79) positively correlated with AP (P < 0.01). Nine taxa, including Chrysosporium (r = 0.84), Cheilymenia (r = 0.83), Podospora (r = 0.79), and Penicillium (r = 0.77), positively correlated with AK (P < 0.01). Penicillium (r = −0.70) was negatively correlated with TN (P < 0.01).
Total nitrogen and potassium explained 20% of microbial variation. Agromyces and Solirubrobacter correlated with available potassium, while Mortierella linked to TN/TP trade-offs. The high-yield variety exhibited stronger positive correlations with AP/AK, reflecting optimized nutrient-microbe synergy. This study highlights the complex interplay between nutrient dynamics and microbial communities in two ginger cultivars, providing valuable insights into plant-microbe interactions in agricultural ecosystems.
Microbial Responses to LPA and Oxindole in In Vitro Assays
The growth dynamics of Devosia riboflavina were significantly modulated by metabolite treatments. Under control conditions, the bacterium displayed a logistic growth pattern, reaching a maximum estimated biomass of 9.61 × 10⁸ CFU/mL. Supplementation with 1-oleoyl-LPA led to dose-dependent increases in final biomass (up to 1.03 × 10⁹ CFU/mL at 50 μM), albeit with modest reductions in growth rate. In contrast, Oxindole induced a biphasic response: 25 μM delayed growth onset (t₀ = 11.35 h) and reduced the rate, while 50 μM significantly enhanced proliferation speed (r = 0.2239 h⁻¹) but decreased final CFU (Fig. 7a).
Fig. 7: Growth dynamics of keystone microbes in response to metabolites and their effects on ginger seedling development.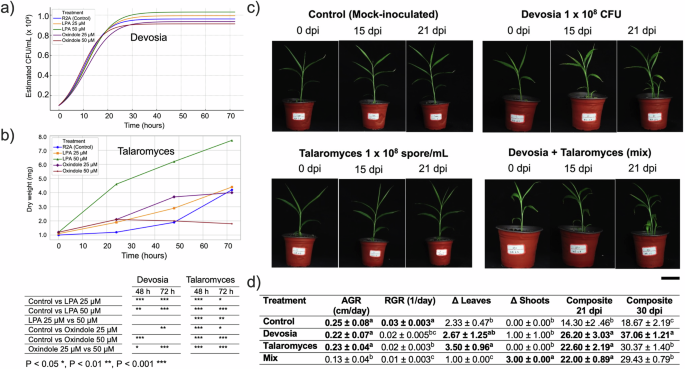
a Growth curves of Devosia riboflavina (OD₆₀₀ converted to CFU/mL) over 72 h under control conditions and in the presence of 1-oleoyl-LPA (25 μM, 50 μM) or Oxindole (25 μM, 50 μM). b Biomass accumulation of Talaromyces pseudofuniculosus (dry weight, mg) under the same metabolite treatments over 72 h. Significant differences were observed at 48 and 72 h. c Representative images of ginger seedlings at 0, 15, and 21 days post-inoculation (dpi) with four treatments: mock-inoculated control, Devosia riboflavina (1 × 10⁸ CFU/mL), Talaromyces pseudofuniculosus (1 × 10⁸ spores/mL), and a microbial consortium (Devosia + Talaromyces). Scale bars, 5 cm. d Morphological parameters measured at 21 dpi, including AGR (absolute growth rate), RGR (relative growth rate), ΔLeaves (change in leaf number from 0 to 21 dpi), ΔShoots (net shoot formation), and the composite growth index integrating shoot number, height, and leaf development at 21 dpi. Composite values at 30 dpi were estimated by linear regression. Data represent mean ± s.d. (n = 6). Asterisks and different letters indicate statistically significant differences (p < 0.05, one-way ANOVA with Tukey’s post hoc test).
Talaromyces pseudofuniculosus exhibited treatment-specific growth responses over the 72-hour incubation period (Fig. 7b). All groups began with comparable baseline dry weights (~1.0 mg at zero h). Biomass increased steadily in all treatments, but the magnitude and kinetics differed significantly. LPA supplementation notably stimulated fungal proliferation in a concentration-dependent manner. At 72 h, LPA 50 μM yielded the highest biomass (7.21 mg), considerably exceeding both the control (3.72 mg) and LPA 25 μM (4.78 mg) treatments (P < 0.05). In contrast, Oxindole exposure repressed growth, with the 50 μM condition showing the weakest biomass accumulation (2.11 mg), followed by the 25 μM group (3.66 mg). The control treatment (R2A without added metabolites) maintained intermediate growth levels. These results confirm that LPA acts as a growth-promoting cue for Talaromyces, while oxindole constrains its expansion, aligning with observed metabolomic associations in the native ginger rhizosphere.
Keystone microbial inoculation shapes ginger seedling development
Microbial inoculation significantly influenced several plant growth traits, including absolute growth rate (AGR), relative growth rate (RGR), leaf and shoot development, and composite biomass accumulation at 21 and 30 days post-inoculation (dpi) (Fig. 7c and d). Compared to the control (AGR: 0.25 ± 0.08 cm/day), plants inoculated with Devosia (0.22 ± 0.07 cm/day) and Talaromyces (0.23 ± 0.04 cm/day) exhibited comparable absolute growth rates (p > 0.05). However, co-inoculation with both microbes (Mix) resulted in a significant reduction in AGR (0.13 ± 0.04 cm/day; p < 0.05). A similar trend was observed for relative growth rate, where the mix treatment showed the lowest RGR (0.01 ± 0.003 1/day), significantly lower than all other treatments.
Talaromyces significantly increased leaf number (Δ Leaves: 3.50 ± 0.96), outperforming both the control (2.33 ± 0.47) and mix treatments (1.00 ± 0.00). Interestingly, the mixed treatment induced the highest shoot proliferation (Δ Shoots: 3.00 ± 0.00), suggesting a potential shift in resource allocation or developmental pattern. At 21 dpi, all inoculated treatments significantly outperformed the control in composite biomass, with Devosia leading (26.20 ± 3.03), followed by Talaromyces (22.60 ± 2.19) and Mix (22.00 ± 0.89). By 30 dpi, Devosia-inoculated plants sustained the highest biomass (37.06 ± 1.21), while Talaromyces (30.37 ± 1.40) and Mix (29.43 ± 0.79) showed intermediate performance. The control remained significantly lower (18.67 ± 2.19).
These results suggest that individual microbial strains (Devosia and Talaromyces) can promote specific aspects of plant growth, with Talaromyces enhancing foliar development and Devosia contributing to sustained biomass accumulation. In contrast, co-inoculation did not synergise benefits and instead reduced growth rate, highlighting the importance of compatibility and functional precision in microbial consortia design.