
Researchers from Finland’s University of Jyväskylä have combined quantum-mechanical modeling and universal machine learning to reveal how geometry determines the stability of graphene – metallene interfaces – an important step toward bringing these promising 2D materials into real-world technologies.
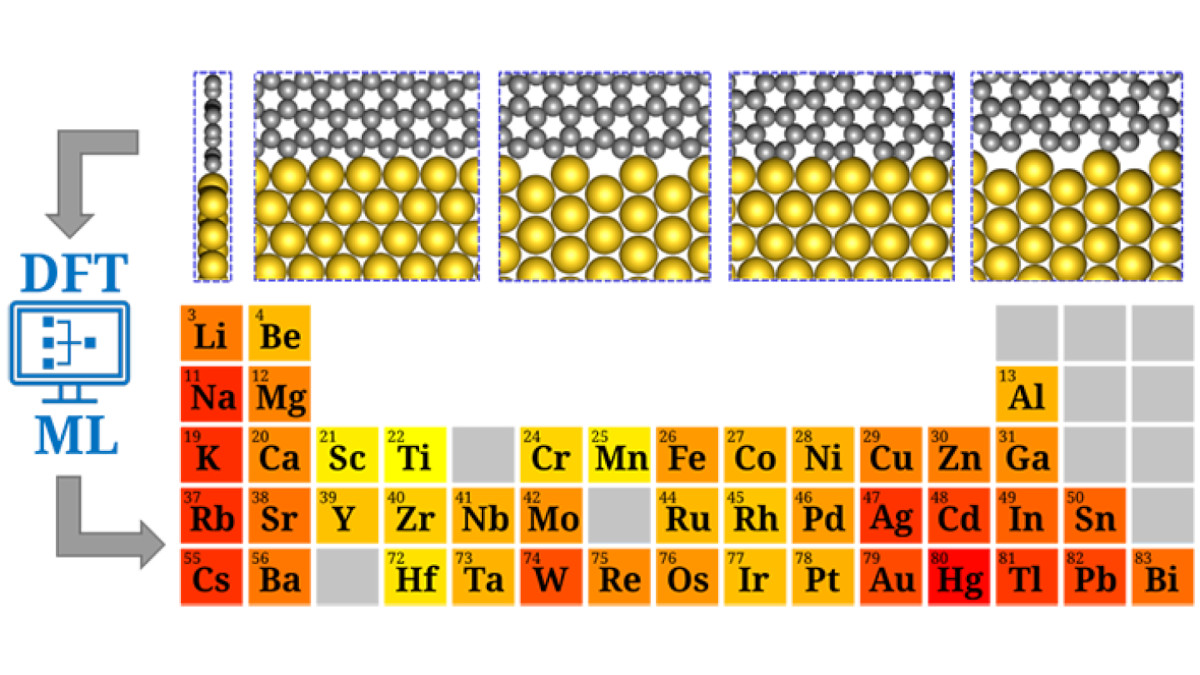
Integrating density-functional theory and machine-learning to assess the stability of lateral graphene–metallene interfaces. Image credit: University of Jyväskylä
Metallenes are atomically thin, nonlayered metallic sheets with great potential for applications ranging from next-generation electronics to energy storage and catalysis. Yet their strong metallic bonding makes them unstable in isolation, often requiring confinement within the pores of 2D templates such as graphene. To tackle this challenge, Professor Pekka Koskinen’s team conducted a large-scale computational study of 1,080 graphene–metallene interfaces, covering 45 different metals and four interface geometries. Using density-functional theory (DFT) alongside the MatterSim machine-learning interatomic potential, the researchers optimized interface structures, analyzed their electronic properties, and tested their stabilities under strain, defects, and thermal motion.
Their results show that smooth, well-aligned geometries – for example, where the zigzag edge of graphene meets a straight metallene edge – lead to the most stable, defect-resistant, and energetically favorable interfaces. In contrast, ragged or mismatched boundaries destabilize the metallic layer, causing reconstruction or collapse. Transition-metal metallenes, in particular, form the most robust and resilient junctions.
Postdoctoral researcher Mohammad Bagheri, who carried out the simulations, said: “We found that interface stability depends on maintaining smooth, well-aligned geometries. Such clean edges provide strong resistance to defects and mechanical strain, whereas irregular boundaries promote destabilization”.
Beyond these physical insights, the study also validates machine learning as a reliable and efficient tool for predicting complex atomistic behaviors, enabling rapid exploration of material combinations that were once computationally prohibitive.
This work establishes fundamental design rules for stabilizing metallenes with graphene and provides geometric and elemental guidelines to accelerate experimental synthesis. As Koskinen notes, “understanding the microscopic principles of interface stability is the key step toward scalable, high-performance metallene materials”. By bridging AI-driven materials modeling and quantum-level physics, this research advances the roadmap for integrating metallenes into practical devices in electronics, energy conversion, and biomedicine.