
Bacterial strains and growth conditionsBacterial strains
The following strains were from the Keio collection1 (Horizon Discovery): E. coli K12 BW25113 WT, ΔompF, ΔompC, ΔompG, ΔnmpC, ΔphoE, Δkch, ΔclcB, ΔptsH, ΔaceA, ΔgtlD, ΔycgG, ΔyidA, ΔushA and ΔtolC. The long-term stock was stored in 20% glycerol at −80 °C. Bacterial samples were stroked monthly in Luria Broth (LB) + agar with or without antibiotics in which kanamycin was used for selection at 50 µg ml−1. Single colonies were resuspended in a 50 ml falcon tube with 10 ml fresh media as indicated. These cultures were incubated in glass test tubes with a breathable lid in a shaking incubator at 37 °C in an orbital shaker at 300 r.p.m.
Media composition
The super-optimal broth (SOB) or minimal medium 9 (M9) was used for each experiment. The SOB was prepared following a premix recipe (Fromedium, SOB01CFG). To prepare the minimal medium, we adapted the recipe described by Kotte2: The medium contained the following components: base salt solution (211 mM Na2HPO4, 110 mM KH2PO4, 42.8 mM NaCl, 56.7 mM (NH4)2SO4, autoclaved and prepared by the scientific facilities at the MRC Laboratory of Molecular Biology (LMB)), 10 ml of trace elements (final concentration: 0.63 mM ZnSO4, 0.7 mM CuCl2, 0.71 mM MnSO4, 0.76 mM CoCl2, autoclaved), 0.1 ml 1 M CaCl2 solution (autoclaved, prepared by the scientific facilities at LMB) for a final concentration of 0.1 mM, 1 ml 1 M MgSO4 solution (autoclaved, prepared by the scientific facilities at LMB) for a final concentration of 1 mM, 2 ml of 500× thiamine solution (1.4 mM in ultrapure water from Milli-Q, Millipore and filter sterilized) and 0.6 ml 0.1 M FeCl3 solution (filter sterilized). The final volume was adjusted to 1 l with ultrapure water (Milli-Q, Millipore) lab water system (Milli-Q Advantage-10, Millipore). For each batch, the medium was filtered through a 0.22 µm Millipore Stericup and split into 500 ml bottles. Before each experiment, the medium was supplemented with 1 g l−1 casamino acids, 1 mM tryptophan and 0.5 g l−1 glucose unless the contrary was specified. Tryptophan stock was prepared at 50 mM in water from powder (Sigma Aldrich) and kept at 4 °C. Casamino acids (VWR Life Science) stock was prepared at 100 g l−1 in MiliQ water and kept at 4 °C.
Carbon sources
The carbon sources used in this work (glucose, fructose, acetate, fumarate, pyruvate) were acquired from Sigma-Aldrich in powder form. Then, stock solutions were prepared at a 100 g l−1 concentration in Milli-Q water. After adjusting the pH to 7.0, 100 ml aliquots were filtrated with a 0.22 µm syringe filter. For the lipid media preparation, we used 14 mg l−1 1,2-dipalmitoylphosphatidylcholine (DPPC), combined with 0.3 g l−1 casitone and 0.05% Tyloxapol to allow proper dilution.
Plasmid design and construction
The primers were designed with the Primer3 algorithm available through the Benchling platform3. The DNA material was amplified by PCR with the PrimeStart HS polymerase kit (Takara), following the manufacturer protocol and adjusting the annealing temperature to the suggested by the Benchling cloning algorithm. The amplification cycles were repeated 30 times using a C1000 Touch Thermal Cycler (BioRad).
The resulting DNA fragments were ligated with the Gibson assembly kit. The concentration of the DNA templates was estimated with UV absorption using a NanoDrop 3300 (ThermoFisher), and the NEBiocalculator helped us estimate the required volumes for the Gibson assembly. All assemblies were carried out for 1 h at 50 °C using the same thermocycler as the PCR amplification. Bacterial transformation of the assembled sequence was carried out through heat shock using E. coli DH5α as an intermediate strain. Isolated plasmids were stored at −20 °C in Qiagen Elution Buffer.
pBAD_pelBCpHuji assembly
The periplasmic pH sensor was based on the arabinose-inducible pBAD TOPO cloning vector system. This backbone was amplified with primers listed in Supplementary Table 1. To this backbone, we added the pectate lyase B (pelB) leader sequence. The pelB export signal sequence has proven to be successful in delivering recombinant proteins to the periplasm of E. coli4,5. This signalling sequence consists of 22 amino acids placed at the start codon. The pelBC fragment was amplified with primers listed in Supplementary Table 1. Then, we inserted the pH sensor. To measure the periplasmic pH accurately, we looked for a sensor with a wide dynamic range because the periplasmic pH could be up to 2 units lower than the cytoplasm, which means a pH 5 (ref. 6,7). As the flurescence of the pH sensor pHluorin collapses below pH 6, we opted for pHuji with a working range between 5 and 9 (ref. 8). For the amplification of the pHuji fragment, we used primers listed in Supplementary Table 1. During the assembly process, we used E. coli DH5α, and after validating the sequence, we transferred the construct to an E. coli K12 BW25113 WT strain.
pBAD_pelBCginko2 assembly
We use the pelBC-pHuji plasmid to extract the backbone with the periplasmic export signal pelB. As GINKO1 is derived from green fluorescent protein, which is not stable in the periplasm, we designed a new variant based on super folding yellow fluorescent protein (sYFP) for improved periplasmic stability. Thus, we amplified the potassium-binding region of GINKO1 with primers listed in Supplementary Table 1 and inserted it between the 171 and 172 amino acids residues of sYFP. The sYFP was amplified with primers listed in Supplementary Table 1. This strategy was designed following the GINKO1 architecture. The final construct was amplified inside E. coli DH5α, and after validating the sequence, we transferred the construct to the E. coli K12 BW25113 strain.
pBAD_ArchT assembly
We used the pLenti-CaMKIIa-eArch 3.0-EYFP plasmid for the ArchT amplification9,10. For the PCR amplification, primers listed in Supplementary Table 1 were used. The backbone was derived from the pBAD_TOPO plasmid via digestion with the NcoI and PmeI restriction enzymes. Then, both fragments were ligated with the Gibson Assembly Kit.
Other genetic material
The plasmids pBAD_GINKO1 (Addgene code: 113111), pBAD_QuasAr2 (Addgene code: 64134) and GCaM-mRuby (also called pJMK0004, addgene code: 98920) were obtained via Addgene. The pBAD_pHluorin plasmid was developed in previous work10,11.
Mutant generation
E. coli DH5a was used as a maintenance strain for all cloning steps. The ompC mutant allele vector (pBAD backbone) was constructed using a combination of traditional digestion/ligation cloning and Gibson assembly of mutant allele and chloramphenicol resistance cassettes, respectively.
Briefly, the ompC mutant allele (ompC_mut) was synthesized by Thermo Life Technologies with the following amino acid changes from the source sequence (ompC_source): E23A, K27A, D28A, E64A, D69A, D156A, D162A, D289A, K329A. The ompC_mut sequence was then amplified using primers listed in Supplementary Table 1 and cloned into the pBAD vector using the restriction site pair NcoI and PmeI to generate pBAD-ompC_mut. A chloramphenicol resistance cassette was amplified from the plasmid pT2SC using primers listed in Supplementary Table 1 and was subsequently introduced to the pBAD vector using Gibson assembly at the PmeI site, generating the final vector pBAD-ompC_mut-ChlR. An ompC_mut-ChlR fragment was then PCR amplified from pBAD-ompC_mut-ChlR with E. coli chromosomal homology regions using primers listed in Supplementary Table 1. The ompC mutant was then generated using E. coli BW25113 and the ompC_mut-ChlR fragment via λ Red recombinase-mediated allelic replacement as described in ref. 77. Mutants were confirmed by PCR and Sanger sequencing12.
Permeability quantification using flow cytometryTracer uptake
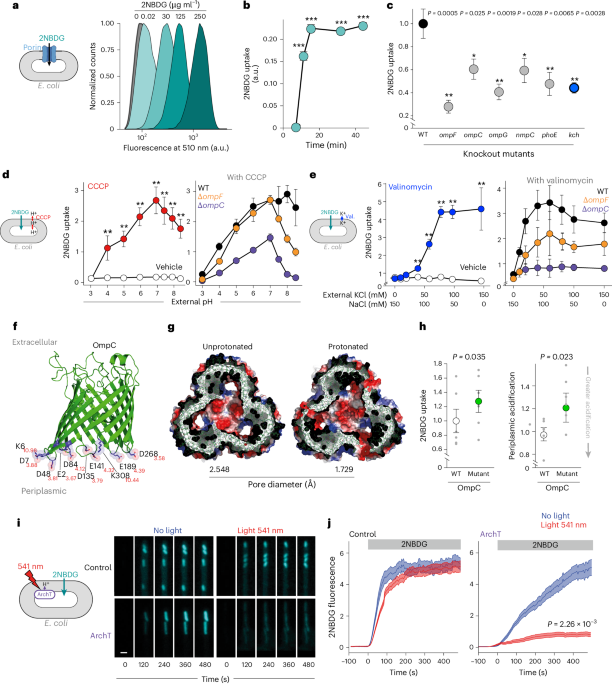
The method for permeability estimation in bacteria was derived from refs. 38,44,78. Briefly, bacteria cultures were grown overnight at 37 °C in minimal medium M9, supplemented with casamino acids at 1 g l−1, tryptophan at 1 mM and glucose at 0.5 g l−1. The next day, the source cultures were diluted to an OD ≈ 0.05 in 25 ml fresh medium and placed in a 250 ml conical flask. Glucose was not added to the medium for 2NBDG experiments. When the turbidity reached an OD of 0.1–0.25, the bacteria were split in a 96-well U-shape plate with 180 μl per well. Here, 10 μl of the treatment solution was added, and the plate was sealed with gas-permeable film (4titude, PCR0548) and returned to the incubator. After 30 min, 10 μl of the fluorescent tracer from the reservoir was added. Then, the concentration of the fluorescent tracer in the reservoir was diluted so we could keep the final volume constant.
After adding the fluorescent probe, the plate was returned to incubation for the specified time. Next, 200 μl was transferred to a V-bottom 96-well plate (Costar), and it was centrifuged 5 min at 3,200 × g (Eppendorf 5810 R, rotor S-4-104) and washed twice with PBS. Finally, samples were fixed and resuspended in PBS + 4% formaldehyde, stored at 4 °C and analysed after 16 h. Then, we used the iCyt Eclipse (Sony) flow cytometer to read the fluorescence from all the samples. This device is equipped with 488 nm, 561 nm and 642 nm laser sources, along with Hoechst and FITC filter sets.
For the data analysis, background fluorescence was subtracted, and the cell size effect was corrected by dividing tracer fluorescence by the forward scatter signal79. For each experiment, the signal was normalized to WT strain or vehicle treatment.
Tracer uptake in different carbon sources
For the permeability estimation in different carbon sources, bacteria were grown as described before. When the cultures’ turbidity reached an OD of 0.1–0.25, the cultures were spun down for 5 min at 3,200 × g (Eppendorf 5810 R, rotor S-4-104), washed with M9 and resuspended in 15 ml fresh medium. The different medium compositions were M9 + casamino acids at 1 g l−1, tryptophan at 1 mM and glucose at 0.5 g l−1; M9 + 4 g l−1 (high) or 0.04 g l−1 (low) glucose; or M9 only (no carbon source). Next, the cultures were transferred to a 37 °C water bath, and 400 μl samples were taken at the indicated time points. A 400 μl sample was transferred to an Eppendorf tube, and 4 μl Hoechst (10 mg ml−1) was added to a final concentration of 10 μg ml−1. The tubes were inverted 3 times and incubated at 37 °C for 10 min. The samples were centrifuged for 30 s at maximum speed at 4 °C (Eppendorf centrifuge 5415 R), washed once and resuspended in 400 μl ice-cold PBS. Data analysis was performed as described in the previous section.
Microfluidics imagingMother machine design and assembly
We used the mother machine design described in refs. 55,80. The microfluidic device was fabricated from an epoxy master template courtesy of Dr Jehangir Cama (University of Cambridge/University of Exeter, UK). The mother machine consists of a feed trench (50 µm × 100 µm × 30 mm) with many channels (1.4 µm × 1.4 µm × 25 µm) attached perpendicular to the trench.
For each batch of 12 chips, 50 ml of poly(dimethyl siloxane) (PDMS) was mixed with a 5 ml curing agent (10:1) with vigorous stirring. The bubbles formed during the mixing were removed by vacuum degassing for 20 min until all air bubbles disappeared. Then, the gel was poured onto the master template and baked at 100 °C for 1 h. Subsequently, the chip was cut out around the wafer and prepared for bonding with the cover slide. Holes for inlets and outlets were punched using a sharpened 0.6 mm biopsy puncher (from Fisher Scientific), and the chip was cleaned with Scotch tape and 2-propanol. After drying all the excess isopropanol, the coverslip and PDMS with the features upwards were exposed to air plasma in a vacuum for 20 s at 0.4 mbar oxygen (PlasmaPrep2, Gala Instrumente). This process activates the PDMS, which was put on a glass coverslip (24 mm × 50 mm, thickness 0.17 ± 0.005 mm, Carl Roth). Finally, the chips were incubated overnight at 65 °C.
Once the chip was ready, we flushed the chamber with 2-iso-propanol and sonicated the chip for 30 min to remove all the debris (FB50, Fisher-Scientific). Then, we coated the internal chip surface to facilitate bacterial attachment with a passivation buffer (containing herring sperm DNA and bovine serum albumin). The passivation buffer contained 10 mg ml−1 of bovine serum albumin and 10 mg ml−1 of salmon sperm in a 3:1 ratio55. The process was carried out overnight. Finally, the chips were stored in a dry cupboard for a week until required.
Trapping cells in the mother machine
The bacteria preparation started with an overnight culture in the specified medium. Before injecting the cells into the microchip, 2 ml of cells was washed by centrifugation (Eppendorf 5810 R) and resuspended in 2 ml of fresh medium. These cells were incubated for 10 min after and then centrifuged and concentrated to 100 µl. This high-density culture was injected into the microfluidic chip and set in a plate shaker at 37 °C for 10 min at 300 r.p.m. (Grant-bio, PHMP). Once the cells were trapped in the channels, the chip was connected with an inlet and outlet tubing (Tygon ND-100-80 Medical Tubing, 0.5 mm ID, 1.52 mm OD). Next, we flushed the chip with plain M9 for 10 min so that any cells remaining in the main channel were removed.
2NBDG uptake
2NBDG permeability was estimated by measuring the increase of fluorescent signal of E. coli cells trapped in the mother machine side channels. The recording set-up of the 2NBDG videos was done in a Nikon-N STORM, with a CCD camera (Andor-DU-897) and a ×100/1.49NA lens. The microscope had a closed environment chamber to maintain the temperature constant at 37 °C. For illumination, we used a 488 nm and 561 nm laser light (Agilent Technologies, MLC-400B). The 2NBDG fluorescence signal was captured with a dichroic mirror 525/50 (TRF49909), and for the 561 nm laser excitation experiments, we used a quad-band filter (97335) in combination with a 525/50 nm filter.
Ciprofloxacin uptake
Cells were grown overnight in M9 + 1 g l−1 casamino acids + 0.5 g l−1 glucose + 1 mM tryptophan. The next day, cells were trapped in the mother machine as described in the previous section. Then, cells were resuscitated for 1–2 h until growth started. Then, 12.5 µg ml−1 of ciprofloxacin was added to the medium and started flowing into the chamber. Ciprofloxacin uptake was measured in a Zeiss 780 microscope, equipped with a UV light source (DPSS 355 nm 60 mW, Coherent), which allowed ciprofloxacin imaging. This instrument was also equipped with a thermal isolation box to keep the temperature constant at 37 °C. The imaging was done with a ×63/1.4NA oil lens. Ciprofloxacin fluorescence detection was done with a modified DAPI filter setting (435/65 nm)15,29.
Ion sensor calibration
Cell trapping was done as described above. Cells were then resuscitated with M9 + 0.5 g l−1 glucose + 1 g l−1 casamino acids + 1 mM tryptophan for 90 min. Then, for pH calibrations, we switched the medium to M9 + 1 g l−1 glucose for 30 min and finally to PBS calibrated to a determinate pH with 1 g l−1 glucose. The pH selected points were 8.5 and 5.5. The same procedure was done by adding 250 µM CCCP to the medium to permeabilize the cells. In the case of the potassium calibration, we used a medium HEPES pH 7.4 with complementary concentrations of KCl and NaCl to keep the osmolarity constant at 150 mM. To permeabilize cells to the extracellular potassium, we treated cells with 100 µM valinomycin. The flowing conditions were kept at 0.15 ml h−1, and during the switch, we increased the speed to 1 ml h−1 for 15 min. The illumination conditions for each sensor were the following: pelBCpHuji (excitation (Ex), 561 nm; emission (Em), 620/60 nm), pHluorin (Ex, 488 nm; Em, 525/50), ginko1 (Ex, 488 nm; Em, 525/50), pelBCginko2 (Ex, 488 nm; Em, 525/50), GCaMp6f (Ex, 488 nm; Em, 525/50) and QuasAr2 (Ex, 561 nm; Em, 650/long pass (LP) filter).
Ion sensor fluctuations
To observe the ion oscillations, we resuscitated cells with M9 + 0.5 g l−1 glucose + 1 g l−1 casamino acids + 1 mM tryptophan. The flow and temperature were kept constant at 0.150 ml h−1 and 37 °C. After 90 min, we started the recording process, which lasted until cells started dividing. For the data analysis, only 45 min before starting cell division was considered.
Carbon source switches
After trapping the cells into the chip, cells were washed with a plain M9 medium. Once the main channel was clear, the flow was kept constant at 0.15 ml h−1 for 30 min. At this point, we changed the syringe to the required carbon source and increased the flow to 1 ml h−1. After letting the medium flow for 2 min, we started the recording.
Agarose pad experiments
Cells were grown overnight in SOB + 100 µg ml−1 ampicillin supplemented with 0.002 g l−1 arabinose and 20 µM retinal if required. The next day, cells were washed with M9, supplemented with the specified carbon source and resuspended for 10 min. Then, 2 µl was transferred to the agar pad. Before starting the recording, pads were left to dry for 5 min.
Agar pads were prepared on the same day of the experiment and discarded afterward. The gel was composed of a 10 ml M9 medium with 1.5 % (wt/vol) low-melting agarose and the specified carbon source. We then proceeded to dissolve the agarose by heating. Once the mixture was entirely homogeneous, the liquid was split into 3 ml portions onto a 35 mm petri dish (Falcon, ref: 353001). These plates were left to dry for 30 min. Once the agar solidified, we used a 6 mm biopsy puncher (Uni-Core, Harris UK) to cut out a single-use disk. We transferred 1 µl of cells on top of these agar disks, and after the drop was absorbed, the pad was moved onto a CELLview dish with glass bottom (627870, Greiner). Recording started immediately.
Minimal inhibitory concentration and EC50 measurements
Minimal inhibitory concentrations (MICs) and effective concentration 50 (EC50), defined here as the drug concentration that induces a 50% maximal inhibitory effect on cell growth81, were determined for E. coli according to the Clinical and Laboratory Standards Institute method M07-A9 (ref. 82). Briefly, E. coli strains were grown to an optical density (A600 nm) of 0.2–0.3 in liquid culture, and 1 × 105 bacteria were added to each well of 96-well plates containing serial dilutions of the antibiotic in triplicate wells per condition and incubated at 37 °C until growth was seen in the control wells. Then, the turbidity absorbance at 600 nm was measured with a ClarioStart (BGM LABTECH). Finally, the data were fitted to a dose–response model using a four-parameter logistic regression equation83. For the experiments in different carbon sources, cultures were grown in M9 + 4 g l−1 glucose or M9 supplemented with lipids (see ‘Tracer uptake in different carbon sources’ for full details).
Growth curves in the presence of ciprofloxacin
Individual E. coli colonies (WT, ΔptsH and ΔaceA) were resuscitated in 10 ml of M9 media supplemented with 1 g l−1 glucose, M9 + lipid media (see ‘Bacterial strains and growth conditions’ for detailed composition), or mixed medium into 50 ml Falcon tubes. These cultures were incubated overnight at 37 °C with shaking. Note that the cultures grown in lipid media were incubated for slightly longer time due to their slower growth rate in this carbon source. Next, the cultures were grown to an optical density (OD650) between 0.2 and 0.3. Finally, these cultures were diluted at 1:1,000, and McFarland unit readings were taken at 1, 2, 4, 8, 16 and 24 h.
Plasmid design and construction
The plasmids presented in this work were constructed using Gibson assembly84. The plasmids pBAD_QuasAr2 (QuasAr2, 64134), pBAD_Ginko1(ginko1, 113111) and pKL004 (GCaMp6f, 98920), were obtained from the Addgene database52,58,59,62. The plasmids for the expression of the pH sensor pHluorin were developed in previous work85. Finally, the plasmids pBAD_ArchT, pBAD_pelBCpHuji and pBAD_pelBCginko2 were developed specifically for this work. Further information on plasmid construction can be found in Supplementary Table 1.
Molecular dynamic simulations
All simulations were performed by GROMACS v.4.6 (www.gromacs.org) with CHARMM3686 force fields for 100 ns. The trimeric OmpC (PDB ID: 2J1N, resolution 2.0 Å, with 346 amino acid residues87) and OmpF (PDB ID: 2OMF, resolution 2.40 Å, with 340 amino acid residues88) were used in this experiment. Eight independent simulations were performed, four for OmpC and four for OmpF. All systems were prepared using the CHARMM-GUI web interface. First, OmpC simulation was run at physiological pH. The second OmpC simulation was run with protonated L3 loop residues (D99, D105, D113, D118, E119) and D315 on the beta-barrel wall behind the L3 loop. The third simulation was run with protonated periplasmic side chains (E2, D6, D12, E43, D48, D84, D135, D141, D187, E189, D228, D268). Protonation state was calculated using the PROPKA3 (ref. 89). The last simulation was run with protonated L3 loop/periplasmic residues. The same hypothesis was applied to OmpF. The first simulation was run at physiological pH. The second OmpF simulation was run with protonated L3 loop residues (D107, D113, E117, E121, D126, D127) and D312 on the beta-barrel wall behind the L3 loop. The third simulation was run with protonated periplasmic side chains (E2, D6, D12, E48, D54, D92, D149, E183, D221). The orientation of proteins in membranes server was used for the orientation and position of the protein in the membrane, and each system was embedded in a pre-equilibrated neutral zwitterionic lipid phosphatidylcholine (POPC) bilayer. All the simulations were achieved at constant pressure (1 atm) and temperature (300 K). The results were analysed by locally written code. This was calculated from the output of multiple sampled conformers. Molecular graphic images were prepared using pymol 3.1.
The porin diameter was measured using the HOLE (v.2.2)90 software. More precisely, the diameter was determined using the block averaging method. The trajectory data were divided into blocks of frames, and the pore size was calculated for each block. The mean and standard deviation of the pore size across all blocks were computed. The standard deviation of the mean over blocks was used as a measure of the statistical error.
Membrane voltage spike frequency
The QuasAr2 spike count was modelled with the glmmTMB91 R package (v.1.0.0). The model considers the concentration of the carbon sources (in log scale) and corrects for the non-spiking cells. The model parameters can be found in Supplementary Table 2.
Statistical analysis
Statistical analyses were performed using R software (version 3.4.0). Data are presented as mean ± standard error of the mean (s.e.m.) from at least three independent experiments performed in triplicate unless otherwise stated. For flow cytometry experiments, background fluorescence was subtracted, and signals were normalized to WT strain or vehicle treatment controls. Statistical significance between two groups was determined using two-tailed Student’s t-test. For non-parametric data, Wilcoxon signed-rank test was applied. Multiple comparisons were corrected using Bonferroni adjustment where appropriate. The frequency of membrane voltage action potentials was analysed using generalized linear mixed models with the glmmTMB package, accounting for concentration effects in log scale and correcting for non-spiking cells. Dose–response curves for antibiotic susceptibility were fitted using four-parameter logistic regression. Statistical significance was defined as P < 0.05, with significance levels indicated as *P < 0.05, **P < 0.01, ***P < 0.001.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.