
Bacterial strains, plasmids, and growth conditions
The bacterial strains, plasmids and all primers used in this study are listed in Supplementary data 1 and Supplementary data 2 in Supplementary file. The E. coli and P. aeruginosa MPAO1 wild-type and mutant strains were grown in LB medium (Bertani, 1951), at 37 °C except where indicated. Carbenicillin (100 µg/mL) was used to maintain pHERD20T-based plasmids and pEX18Ap-based plasmids, chloramphenicol (30 µg/mL) was used to maintain pTac-based plasmids and gentamycin (30 µg/mL) was used to maintain pEX18Gm-based plasmids, which were used to delete genes in MPAO1. 0.3% (w/v) L-arabinose was used to induce the expression of target genes in pHERD20T-based plasmids. 1 mM IPTG (isopropyl-β-D-thiogalactopyranoside) was used to induce the expression of target genes in pTac-based plasmids.
Deletion of genes in the MPAO1 chromosome
The mutant strains were generated using a rapid method for the generation of P. aeruginosa deletion mutants with modifications53. Considering the co-transcription of PA0717-PA0718-PA0719, we only mutated the start codon of each gene and inserted a gentamycin sequence. Fragments were ligated into pEX18Gm/pEX18Ap plasmid digested with EcoRI and HindIII using one-step ligation kit (Vazyme Biotechnology Corporation, Nanjing, China). Then the ligated products were transferred into the diaminopimelic acid (DAP) auxotrophic RP4 conjugal donor E. coli strain WM3064. The correct constructs were confirmed by both PCR and sequencing. Then they were conjugated into MPAO1 cells. Following this, the in-frame deletion mutants were generated via homologous recombination using the sucrose resistance selection method. The mutations were verified using PCR and DNA sequencing with the gene-LF/LR primer pair.
Biofilm growth and sample collection
A flow cell biofilm system cultured in M9 medium (47.8 mM Na2HPO4, 22 mM KH2PO4, 6.8 mM NH4Cl, 18.7 mM NaCl, 100 µM CaCl2, 2 mM MgSO4 and 0.1% glucose) was conducted as previously described with minor modification6. In details, 1 mL of fresh overnight cultures was injected into the inlet of medical silicone catheters (with an inner diameter of 3 mm and a length of 400 mm, produced by Forbest Manufacturing Corporation, Shenzhen, China), taking care to prevent bubble formation. Subsequently, the inoculated catheters were left static for 1 h to facilitate colonization of the bacteria on the inner surface of the catheters. After this colonization period, a peristaltic pump was used to initiate flow at a rate of 0.1 mL per minute for each channel. At specific time points, phages were collected from the effluents, and biofilm cells inside the catheters were obtained by cutting the catheters open. During the experiment, fresh M9 medium was supplied daily, and the total assay runtime varied between 4 and 6 days, depending on the specific readout requirements. Phages in biofilm effluents were collected on mentioned days by collecting 2 mL of newly flowed M9 culture. Subsequently, the collected effluents were filtered through a 0.22-μm pore size filter (Millipore) and treated with 1 µg mL−1 DNase I (New England Biolabs, MA, USA) at room temperature for 1.5 h and stored at 4 °C54. Biofilm cells in the inner surface of the medical catheter were collected by cutting 1 cm section and stored at −80 °C.
Quantification of Pf4 and Pf6 phages in effluents
Total phage titers in the effluents were quantified using the traditional top-layer agar method55. In brief, the treated effluents were serially diluted 10-fold in SM buffer, and a volume of 5 μL medium of each dilution was spotted on R-top layer media supplemented with the MPAO1 wild-type or ΔPf4ΔPf6 strains. Then the agar plates were incubated at 37 °C for 16 h. After that, the phage-forming units in biofilm effluents were calculated by multiplying the number of countable plaques on agar plates by the dilution fold and then by 200. The ratio between Pf4 and Pf6 phages in biofilm effluents was determined using a qPCR assay.
Quantitative PCR (qPCR) assay
The quantification of Pf4 and Pf6 genes in biofilms and planktonic cells was determined using a qPCR-based method we established previously6. For the quantification of Pf phages, a volume of 1 μL stored effluent was treated with DNase I and then used as a PCR template, and the MPAO1 chromosome-specific gene gyrB was used as a negative control to exclude contamination of host genomic DNA. For the biofilms and planktonic cells, genomic DNA was isolated using a TIANamp Bacteria DNA Kit (Tiangen Biotech Co., Ltd., Beijing, China) following the manufacturer’s instructions. During the DNA isolation process, RNA was removed by adding 10 U/μL RNase A during cell lysis. A total of 20 ng of DNA from each sample was utilized to quantify the copy numbers of Pf genes. The fold change of Pf4 and Pf6 in each effluent/cell sample was calculated based on their specific genes listed in Supplementary data 2. For Pf4 phages, specific Pf4 primers (Pf4-attB, Pf4-attP, xisF4, pfiA, and repF4) were used, which are not present in Pf6. For Pf6 phages, specific Pf6 primers (Pf6-attB, Pf6-attP, intF6, pfkA, and repF6) were used, which are not present in Pf4.
Copy numbers of Pf4 and Pf6 phages in biofilm cells
Total genomic DNA was isolated from biofilm cells using a TIANamp Bacteria DNA Kit (Tiangen Biotech Co., Ltd., Beijing, China) following the manufacturer’s instructions. To exclude RNA contamination, the potential RNA was removed by adding 10 U µl−1 of RNase for 20 min at room temperature during the DNA isolation process. Then 20 ng DNA of each sample was used as template. In addition to pfiA and intF6, Pf4-attP was used to determine the copy number of Pf4 and Pf6-attP was used to determine the copy number of Pf6 in the same cells.
Co-expression toxicity assay
Co-expression of KKP was performed using two-plasmid system with pTac and pHERD20T promoters. For measurements of colony-forming units (CFU), overnight cells were diluted to an OD600 ≈of 0.1, and cells were cultured for 6 h in LB medium with 0.1 mg mL−1 carbenicillin and 0.03 mg mL−1 chloramphenicol, followed by being diluted serially and dropped onto the plate with 1mM IPTG and 0.3% L-arabinose. Plates with 0.1 mg mL−1 carbenicillin and 0.03 mg mL−1 chloramphenicol were used as a control. Then, plates were incubated at 37 °C for 16 h.
Whole-genome sequencing of phages and resequencing of hosts
The experiments were conducted strictly following our previous study38. In brief, whole bacterial genomic DNA and whole ssDNA genome of Pf phages in biofilm cells and effluents were isolated using a TIANamp Bacteria DNA Kit and a TIANamp Virus DNA/RNA Kit (Tiangen, Beijing, China). Then the following double-stranded DNA generating from ssDNA, DNA library construction and sequencing are carried out in GENEWIZ (Suzhou, China).
Plaque assay
Pf phages released from biofilms as reported previously were collected at mentioned days56,57. A total of 1 mL of culture was centrifuged, and the supernatant was filtered through a 0.22-μm filter (Millipore Millex GP) to obtain the Pf phage solution. The filtered Pf phage was serially diluted with LB, and 5 μL was dropped onto bacterial lawns. For the overexpression strains, the overnight cultures were diluted to OD = 0.1 and cultured for 2 h, then L-arabinose was added to induce the expression of genes for 4 h. The double-layer agar plates for the plaque assay were prepared as reported previously37.
Protein 3D structure prediction
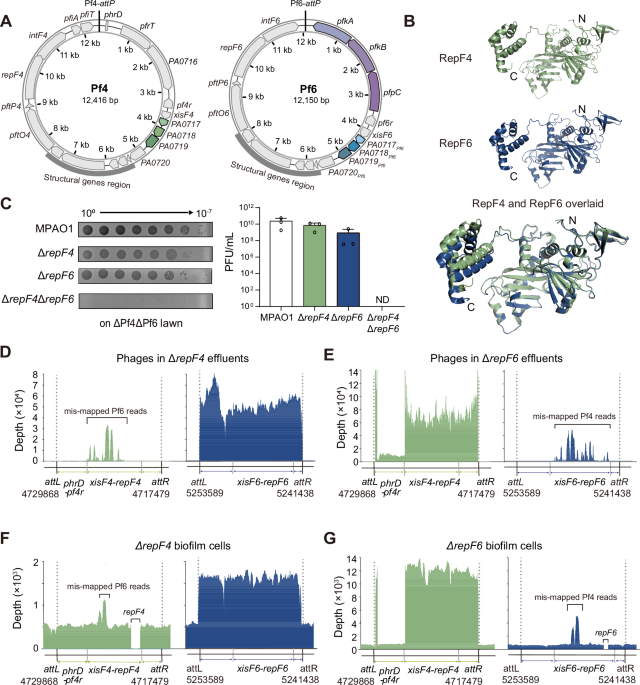
Structure of Pf6r (PDB ID: 6X6F) was based on the crystal structure in PDB. The tertiary structures of RepF4, RepF6, XisF4, XisF6, Pf4r, RepG4 and RepG6 were predicted with AlphaFold2 (https://alphafold.com/)58. For structural alignment, structural figures were produced with PyMOL (www.pymol.org).
Comparison of Pf phages
The Pf phages from different strains were compared by tBLASTx using program Easyfig_2.2.5_win59. In brief, the amino acid sequences of different Pf phages were downloaded from NCBI in GenBank format, followed by uploading to Easyfig_2.2.5_win and being aligned with tBLASTx.
Protein purifications
A 500 mL LB medium containing the indicated antibiotic was inoculated with 3 mL overnight pre-culture and grown at 37 °C with shaking (200 rpm). Inducer was added from OD600 ≈ 0.5 and culture were incubated at 37 °C for 16 h. Overnight cell pellets were collected by centrifugation (4500 × g for 30min) and resuspended using Buffer A (500 mM NaCl, 40 mM Tris-HCl, 10 mM imidazole, pH 8.0), followed by being lysed via ultrasonication, and the supernatant was collected. Ni-NTA resin was used to bind the His-tagged protein, and proteins were eluted with increasing concentrations of imidazole. The eluted proteins were collected, dialyzed against buffer (200 mM NaCl, 20 mM Tris-HCl, pH 8.0) and stored on ice.
Protein degradation assays
Purified proteins were incubated with the cell lysates for the indicated times at 37 °C/4 °C. In detail, overnight cultures were diluted to OD600 ≈ 0.1 and cultured for 8 h. Then, the cell pellets were collected; for overexpression cells, the overnight cultures were diluted to OD600 ≈ 0.1 and cultured for 4 h and then L-arabinose was added to induce target gene expression for 4 h. The cell pellets were resuspended in lysis buffer (200 mM NaCl, 20 mM Tris-HCl, pH 8.0), followed by ultrasonication and centrifugation to collect the supernatant. Equal-volume aliquots of the lysate were then co-incubated with 0.1 μg/μL PfpC for 0, 15, 30, and 60 min at 37 °C, followed by western blotting.
Western blotting
Proteins were separated on Tricine gels, transferred to PVDF membranes (Millipore), and blocked with Fast Blocking Buffer (Yeasen, #36122ES60, China). The membranes were incubated with primary antibodies (His Tag Mouse Monoclonal Antibody, #AF2876, 1:3000, Beyotime; Flag Tag Mouse Monoclonal Antibody, #AF2852, 1:3000, Beyotime; RNA polymerase beta Antibody, #T57097M, 1:3000, Abmart) at 4 °C overnight. After washing with 1 × TBST (diluted from 10 × TBST, Yeasen #60145ES76), the membranes were probed with Secondary Antibody (HRP-conjugated goat anti-mouse IgG(H + L), #A0216; 1:3000, Beyotime; HRP-conjugated goat anti-rabbit IgG(H + L), #A0208, 1:3000, Beyotime) for 1 h at room temperature. Signals were detected using an imaging system (Tanon 5200, China) and the band retention percentage was calculated by ImageJ software (1.57f) and displayed below the corresponding band.
Phosphatase activity assay
The phosphatase activity of PfpC was determined using the Beyotime Alkaline Phosphatase Assay Kit (Nanjing, China) following the manufacturer’s instructions. Purified PfpC was premixed with different concentrations of PA0717 and incubated on ice for 30 min to allow the formation of the complex. The reaction system includes 10 μL of 0.5 μg/μL PfpC (molar mass ~12 μM) as the fixed component, supplemented with 1:1, 1:2, and 1:3 molar ratios of PA0717 (0.1 μg/μL; molar mass ~12 μM). Then, the substrates were added into the reaction system and incubated at 37 °C for 5 min. PfpC phosphatase activity was expressed as nmol min−1 mg−1 protein. Enzymatic activity was measured three times with the same sample.
Pull-down assay
Flag-tagged PfpC was cloned into pETDuet digested with NdeI and XhoI to construct pETDuet-flag-pfpC, and it was then digested by NcoI and HindIII. Next, N-terminal 6xHis-tagged repG4 was introduced to obtain pETDuet-His- repG4-Flag-PfpC. The plasmid was transformed into E. coli strain Rosetta cells, and the cells were cultured at 37 °C. The overexpression was induced by 0.5 mM IPTG at 16 °C for 12 h when the OD600 value reached 0.6–0.8. Then, the pull-downs with the cloned genes were performed. Briefly, pellets were collected by centrifugation at 4500 × g for 30 min at 4 °C. The cell pellet was resuspended using Buffer A (250 mM NaCl, 40 mM Tris-HCl, 10 mM imidazole, pH 8.0) and lysed via ultrasonication, and the supernatant was collected. Ni-NTA resin was used to bind the His-tagged protein, and proteins were eluted with increasing concentrations of imidazole. Rosetta/pETDuet-flag-pfpC was included as a control. The Elute were then subjected to Tricine-SDS PAGE and Western blotting using anti-His and anti-Flag tag antibodies.
Statistics and reproducibility
For all experiments, at least three independent biological replicates were used unless otherwise noted. The experiments were independently repeated twice, yielding similar results. Significance testing was performed by an unpaired T-test for comparisons between two groups, and a one-way analysis of variance (ANOVA) test with Tukey’s correction for multiple comparisons. A p-value of <0.05 was considered statistically significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.