
Serum samples
A total of 100 brucellosis positive serum samples and 96 brucellosis negative serum samples were obtained from the Xuzhou Center for Disease Control and Prevention. All samples were confirmed as positive or negative using the standard tube agglutination test. Additionally, 27 serum samples from patients with fever (non-brucellosis) caused by other pathogens were utilized to evaluate the cross-reactivity of the developed methods.
Preparation of Recombinant T4SS proteins
The amino acid sequences of the VirB proteins were selected based on relevant literature17 and the NCBI protein database. The sequences of VirB1, VirB2, VirB5, VirB6, and VirB7 were analyzed using the UniProt website (https://www.uniprot.org/uniprotkb), and transmembrane regions, signal peptides, and hydrophobic regions were removed. Following a comprehensive evaluation, VirB1, VirB5, and VirB6 were expressed using prokaryotic expression systems, while VirB2 and VirB7 were synthesized as peptides (see Table 1).
Table 1 Protein sequence-based analysis and screening of type IV secretion system proteins.Peptide synthesis
Peptide synthesis was outsourced to a professional peptide synthesis company (Beijing Protein Innovation Co., Ltd.). During the synthesis process, reaction conditions—including temperature, reaction time, and reagent concentration—were meticulously controlled. After each synthesis step, the purity of the peptides was assessed using high-performance liquid chromatography (HPLC, Agilent 1260, USA) to ensure a purity level exceeding 90%. The synthesized VirB2 and VirB7 peptides were then conjugated with keyhole limpet hemocyanin (KLH, Sigma) as a carrier. Following conjugation, unreacted reagents and impurities were removed through dialysis to obtain the final VirB2-KLH and VirB7-KLH conjugates.
Prokaryotic expression
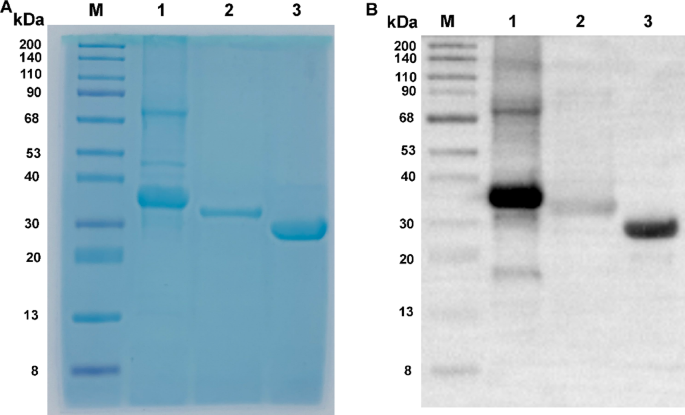
Based on the amino acid sequences of VirB1, VirB5, and VirB6 listed in Table 1, linkers (“GGGS”) were incorporated between adjacent sequences. Codon optimization was conducted to facilitate prokaryotic expression, and a 6xHis tag was appended to the 3’ end for subsequent protein purification. The recombinant protein genes were synthesized by Beijing Protein Innovation Co., Ltd. These synthesized genes were then cloned into the pET30a expression vector and transformed into BL21 cells for IPTG-induced expression. The specific steps were as follows: BL21 competent cells, stored at −80 °C, were thawed on ice and mixed with pET30a(+) before being incubated on ice for 30 min. The mixture underwent a heat shock at 42 °C for 90 s, followed by immediate cooling on ice for 2 min. Subsequently, 800 µL of LB medium (L113084, Aladdin, USA) was added, and the mixture was incubated at 37 °C for 45 min. The cells were centrifuged at 5000 rpm for 3 min, and most of the supernatant was discarded, leaving approximately 100–150 µL to resuspend the cells. The suspension was plated on an LB agar plate containing the 50 µg/mL of Kanamycin and cultured overnight at 37 °C. The cultured bacterial suspension was then transferred to 250 mL of LB liquid medium containing the corresponding antibiotics and incubated at 37 °C with shaking at 200 rpm until the optical density at 600 nm (OD600) reached 0.6–0.8. The cells were induced with 0.5 mM IPTG (16758, Sigma, Germany) at 37 °C for 4 h. Following this, the cells were collected by centrifugation at 8000 rpm for 6 min, and the pellet was resuspended in 20–30 mL of 10 mM Tris-HCl (pH 8.0) solution and sonicated (500 W, 180 cycles, with each cycle consisting of 5 s on and 5 s off). A 100 µL aliquot of the lysed bacterial suspension was centrifuged at 12,000 rpm for 10 min, and 50 µL of the supernatant was transferred to another EP tube. The pellet was resuspended in 50 µL of 10 mM Tris-HCl (pH 8.0) solution. A 12% SDS-PAGE (P0012AC, Beyotime, Shanghai, China) was performed to determine whether the target protein was present in the supernatant or the pellet for subsequent purification.
The nickel column (Ni Sepharose 6 Fast Flow, GE Healthcare) was washed with deionized water until the pH reached 7.0, and then equilibrated with approximately 100 mL of 10 mM Tris-HCl (pH 8.0, T3253, Sigma, Germany). The chromatography column was further equilibrated with about 50 mL of a 10 mM Tris-HCl (pH 8.0) solution containing 0.5 M NaCl (A501218-0001, Sangon Biotech, Shanghai, China). The sample containing the target protein was diluted and loaded onto the column. After loading, the column was washed with a 10 mM Tris-HCl (pH 8.0) solution containing 0.5 M NaCl. Proteins were eluted using 10 mM Tris-HCl (pH 8.0) solutions containing 15 mM, 60 mM, and 300 mM imidazole (with 0.5 M NaCl). Protein peaks were collected and analyzed for purification efficiency using 12% SDS-PAGE. Protein quantification was performed using a BCA protein assay kit (P0010, Beyotime). It is important to note that no dialysis was performed during the purification process, as the proteins were expressed in a soluble form.
The purified protein was verified using Western blot (WB), following these steps: First, the samples were mixed with Loading Buffer at a 1:1 ratio and heated at 95 °C for 5 min to denature the proteins. Next, a 10% SDS-PAGE gel was prepared using the One-Step Fast PAGE Gel Casting Kit (G2177-50T), and the gel was run at 200 V for 35 min in SWE Rapid High-Resolution Electrophoresis Buffer after loading the protein samples and Prestained Protein Marker IV (8-200 kDa, G2083-250UL). Following electrophoresis, the gel was transferred to a membrane using Ice-free Rapid Transfer Buffer at a constant current of 400 mA for 25 min. The membrane was then washed three times with TBS-T for 5 min each. Afterward, the membrane was blocked with 5% skim milk in TBS-T at room temperature for 1 h and washed again with TBS-T three times for 5 min each. Subsequently, the membrane was incubated with Recombinant Anti-His Tag antibody (Mouse mAb, GB151251-100) diluted according to the manufacturer’s instructions (typically 1:1000-1:5000) at 4 °C overnight or at room temperature for 2 h, followed by three 5-minute washes with TBS-T. Next, the membrane was incubated with HRP-conjugated Goat anti-Mouse IgG (typically 1:5000-1:10000, GB23301) at room temperature for 1 h and washed three times with TBS-T for 5 min each. Finally, the membrane was placed in Substrate Solution for color development in the dark for 5–10 min, and the reaction was terminated with water to observe the bands. All reagents were purchased from Wuhan Servicebio Technology Co., Ltd (China).
Establishment of indirect ELISA for serum detection
The indirect enzyme-linked immunosorbent assay (iELISA) was performed as follows: Purified proteins and synthetic peptides were diluted to a concentration of 10 µg/mL in carbonate buffer solution (CBS, pH 9.6), and 100 µL of this solution was added to each well of a 96-well microplate (Corning, USA). The plate was incubated overnight at 4 °C. After washing the wells three times with PBST, 300 µL of blocking solution (5% skim milk in PBS) was added to each well and incubated at 37 °C for 2 h. The plate was washed again with PBST, and human serum diluted in PBS (1:200) was added, followed by incubation at 37 °C for 1 h. After three additional washes with PBST, 100 µL of HRP-conjugated rabbit anti-human IgG (diluted 1:10,000, A18903, Thermo Fisher Scientific, USA) was added to each well and incubated at 37 °C for 1 h. The plate was washed three times with PBST, and tetramethylbenzidine (TMB, T2573, TCI, Japan) substrate solution was added, followed by incubation in the dark for 10 min to allow color development. The reaction was terminated with 2M H2SO4, and the optical density at 450 nm (OD450) was measured using a microplate reader (Versa Max Microplate Reader, Molecular Devices, USA).
In the iELISA assay, the positive controls used were serum samples from patients previously confirmed to be positive for brucellosis by standard tube agglutination tests. The negative controls were serum samples from healthy individuals who had no history of brucellosis or exposure to Brucella spp. Additionally, LPS (provided by the Chinese Animal Health and Epidemiology Center, 3 mg/mL) served as a positive antigen control. Each serum sample was measured in triplicate using the same procedure. Sensitivity, specificity, area under the curve (AUC), and cut-off values were determined through receiver operating characteristic (ROC) analysis.
Assessment of Cross-Reactivity via indirect ELISA
The cross-reactivity of the Brucella T4SS recombinant proteins was assessed using serum samples from patients with fever who did not have brucellosis, in comparison to LPS. Cross-reactivity was evaluated via iELISA as above and based on the cutoff values determined by the ROC curve.
Statistical methods
Dot plots and ROC curve analyses were performed using GraphPad Prism version 6.05. Statistical analyses were conducted using unpaired Student’s t-tests, with a significance level set at P < 0.05.