
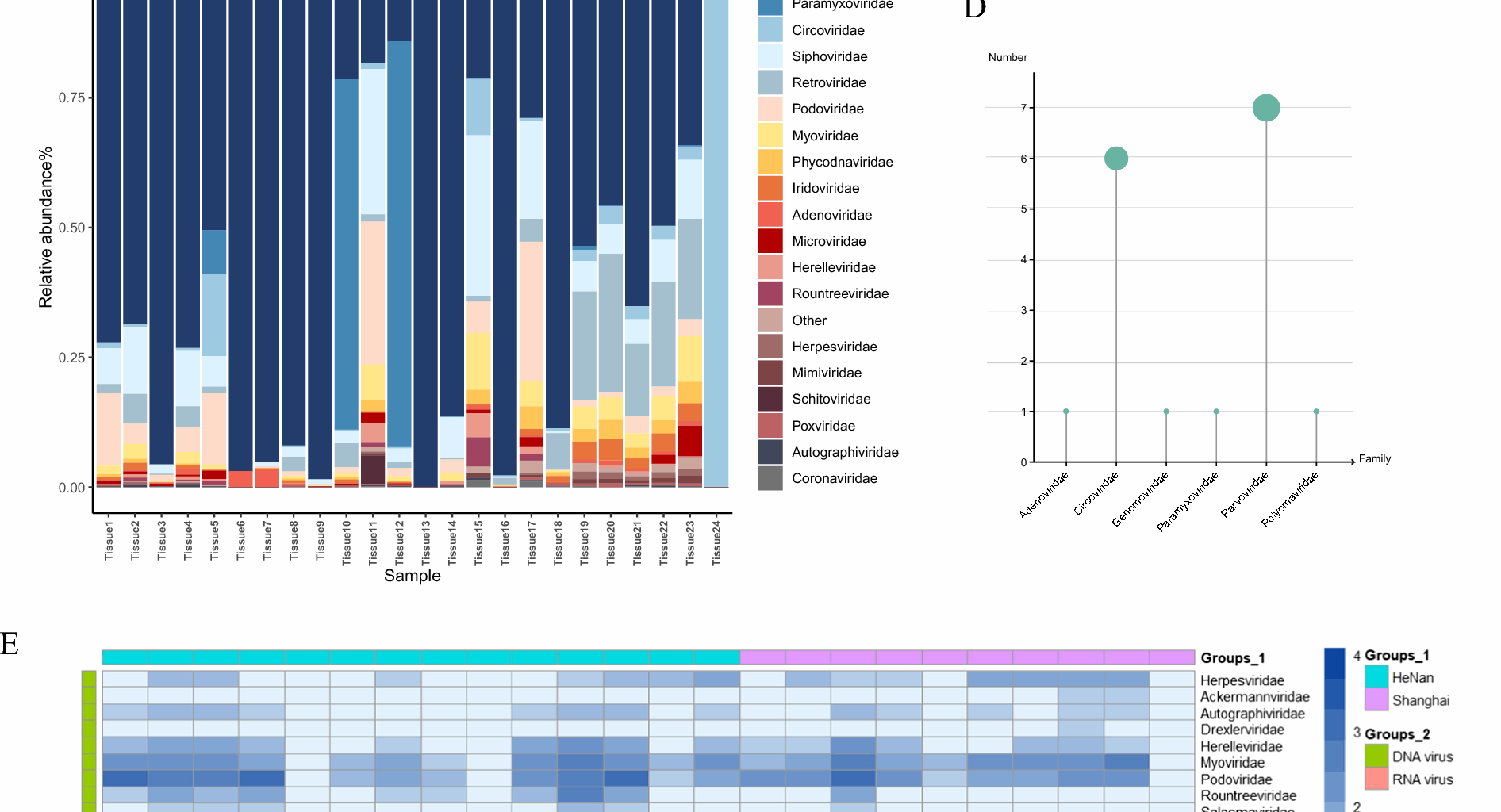
Overview of the virome
To gain a deeper understanding of the viral communities in canine lymph node tissues, this study collected 24 lymph node tissue samples from dogs in close contact with humans in Shanghai and Henan. These samples were subjected to next-generation sequencing on the Illumina NovaSeq platform, generating 24 nucleic acid libraries. A total of 1,854,862,388 raw sequences were obtained from the 24 libraries and after data processing and alignment against the Non-Redundant Protein Sequence Database using BLASTx (E-value < 10–5), 32,969,131 viral reads were identified. The rarefaction curves plateaued, indicating that the sequencing depth of the 24 libraries in this study was sufficient to reflect the species diversity within the samples. Further increases in sequencing depth would not yield additional species (Fig. 1A). To further validate the comprehensiveness of viral detection, species accumulation curves were analyzed. These curves plateaued when the number of viral species exceeded 200, suggesting that the detection of viruses in the study samples had reached saturation (Fig. 1B).
Fig. 1
Viral diversity and taxonomic distribution in 24 canine metagenome libraries. A Rarefaction curves showing species richness across samples (MEGAN v6.21.16). B Species accumulation curves with boxplots representing sample richness; shaded area indicates 95% confidence interval. C Relative abundance of the top 20 viral species across libraries. D Counts of complete viral genomes assembled per viral family. E Heatmap of normalized read counts per viral family in each sample pool
To demonstrate the differences in viral composition within the virome of the samples, we analyzed the identified reads, grouping them by sampling location and nucleic acid type. In 18 out of the 24 libraries, Parvoviridae exhibited the highest relative abundance, while Circoviridae showed the highest relative abundance in the 24th library (Fig. 1C). Furthermore, through data assembly, we obtained complete genome for the Adenoviridae (n = 1), Circoviridae (n = 6), Genomoviridae (n = 1), Paramyxoviridae (n = 1), Parvoviridae (n = 7) and Polyomaviridae (n = 1) (Fig. 1D). Furthermore, we found that the majority of viruses identified in this study showed high sequence homology with known viruses (Table 1). Among the DNA viral families, the highest number of reads was identified for the Parvoviridae (n = 16,641,746), followed by the Circoviridae(n = 3,805,457), Adenoviridae (n = 440,104), Siphoviridae (n = 322,417), and Podoviridae (n = 259,495). According to the International Committee on Taxonomy of Viruses (ICTV), these families, including Parvoviridae, Circoviridae, and Adenoviridae, are known to infect vertebrates. Among the RNA viral families, we identified the Paramyxoviridae (n = 268,349), Retroviridae(n = 59,284), Coronaviridae (n = 1,986) and Picornaviridae (n = 1,313), all of which are also capable of infecting vertebrates (Fig. 1E). Through viral macrogenome sequencing, we identified 39 virus families at the family level. The detailed sequence reads number statistics of each virus family are detailed in Supplementary Table 2.
Table 1 Assembled sequences with identity to those of previously described virusesAdenoviridae
Adenoviridae is a family of non-enveloped viruses with a double-stranded linear DNA genome, which can be classified into six genera: Mastadenovirus, Aviadenovirus, Atadenovirus, Ichtadenovirus, Testadenovirus, and Siadenovirus. Among these, Mastadenovirus primarily infects mammals. The major capsid proteins include the Hexon protein (forming the main capsid structure), the penton protein, and the fiber protein (mediating viral attachment). Notably, the Hexon protein is highly conserved [30, 31]. The adenovirus genome ranges in length from 26 to 48 kbp and encodes early (E1-E4) and late (L1-L5) transcriptional units, which are crucial for viral DNA replication. Adenoviruses can be transmitted through respiratory droplets, the fecal–oral route, and direct contact [32]. They are known to cause respiratory and gastrointestinal infections. In immunocompromised individuals, adenoviruses can lead to severe conditions such as hemorrhagic cystitis and hepatitis [33,34,35].
In this research, in addition to assembling a complete genome of 30,624 bp (C_AA105018), a partial sequence of 14,201 bp (C_AA105027) containing the complete Penton ORF was also assembled. The Penton ORF of this partial sequence showed 87.8% identity with the complete genome. To investigate the relationship between the sequences identified in this study and other adenoviruses, phylogenetic trees were constructed using the highly conserved hexon and penton proteins, respectively (Fig. 2). Both trees indicated that the sequence belongs to the Mastadenovirus genus, which primarily infects mammals. Canine adenovirus (CAV) has two types: Canine adenovirus type 1 (CAV-1), which causes infectious canine hepatitis, and Canine adenovirus type 2 (CAV-2), primarily associated with respiratory disease in dogs. In recent years, the incidence of canine adenovirus infections has been increasing [36, 37]. BLASTx analysis revealed that Tissue7_C_AA105018 exhibited 100% sequence homology with the canine-derived reference sequences for Canine mastadenovirus A strain Toronto A26/61 (NC_001734) and strain NWT-W85 (OK546121). The latter strain (NWT-W85) was isolated from Canis lupus in the Northwest Territories, Canada. Phylogenetic analysis based on Hexon protein demonstrated that the Adenoviridae sequences obtained in this study form a monophyletic branch together with adenoviruses isolated from a variety of mammalian hosts, including bears, sea lions, deer, and humans, suggesting that these viruses may share a common evolutionary ancestor, highlighting the potential risk of infection to humans.
Fig. 2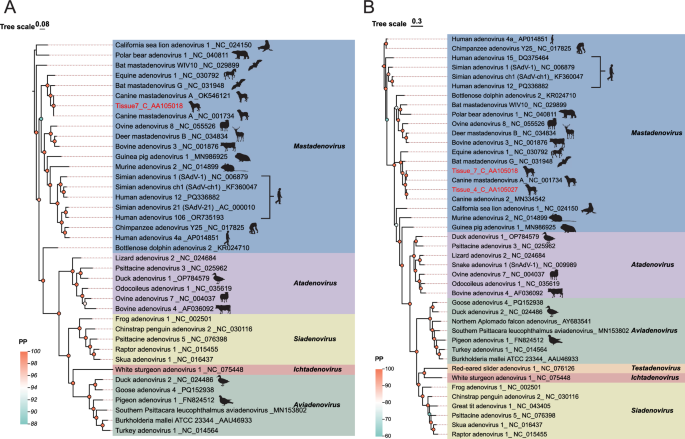
Phylogeny of Adenoviridae based on Hexon and Penton proteins. A Hexon protein tree. B Penton protein tree. Sequences identified in this study are highlighted in red. Scale bars indicate evolutionary distance
Paramyxoviridae
The Paramyxoviridae family consists of single-stranded, negative-sense RNA viruses. According to the latest classification (15th ICTV Report, 2023), this family is divided into four subfamilies: Avulavirinae, Metaparamyxovirinae, Orthoparamyxovirinae, and Rubulavirinae, encompassing 17 genera. These include significant pathogens such as the Morbillivirus and the Orthorubulavirus [38]. Paramyxovirus cause a wide range of diseases in humans, ranging from self-limiting infections to highly fatal encephalitis. Although diseases like measles and mumps can be controlled through vaccination, emerging pathogens such as Henipavirus still pose significant threats [39,40,41].
The viral family possesses a genome spanning approximately 15–19 kilobases, which encodes six canonical open reading frames (ORFs) corresponding to the nucleocapsid (N), phosphoprotein (P), matrix (M), fusion (F), hemagglutinin (H), and large polymerase (L) proteins [42]. To date, research has confirmed that the only pathogen from the Paramyxoviridae detected in canine lymph node tissues is the Canine Distemper Virus (CDV), whose natural host range is primarily limited to species within the mammalian orders Carnivora. In line with this established knowledge, our study also identified a paramyxoviral sequence in canine lymph nodes. In this study, a complete viral genome sequence of 16,096 bp was assembled from high-throughput sequencing data. Genome annotation revealed that it encodes typical structural proteins of Paramyxoviridae. The L protein (RNA-dependent RNA polymerase) is highly conserved across the family Paramyxoviridae, a property that makes it a reliable molecular marker for delineating subfamilies, as adopted by the ICTV. In contrast, the fusion protein (F protein) exhibits relatively lower conservation compared to the L protein. Its functional role in mediating viral entry renders it subject to persistent selective pressure from the host immune system, consequently accelerating its evolutionary rate [38]. Based on this, a phylogenetic tree was constructed using MCMC method [43] (Fig. 3). Phylogenetic analysis and sequence alignment results classified the target sequence within the Paramyxoviridae, specifically the Orthoparamyxovirinae and the Morbillivirus. BLASTx analysis demonstrated that the sequence exhibited 99.7% identity with a CDV (GenBank accession: PQ860808) originating from Henan Province, indicating high sequence homology. CDV classified within Morbillivirus of Orthoparamyxovirinae, is a pathogenic agent that affects the respiratory, gastrointestinal, and neurological systems of canines. While Canis lupus familiaris serve as primary hosts, accumulating evidence has demonstrated its significant impact on endangered wildlife species, including Panthera leo, Panthera pardus, and Ailurus fulgens [44, 45].
Fig. 3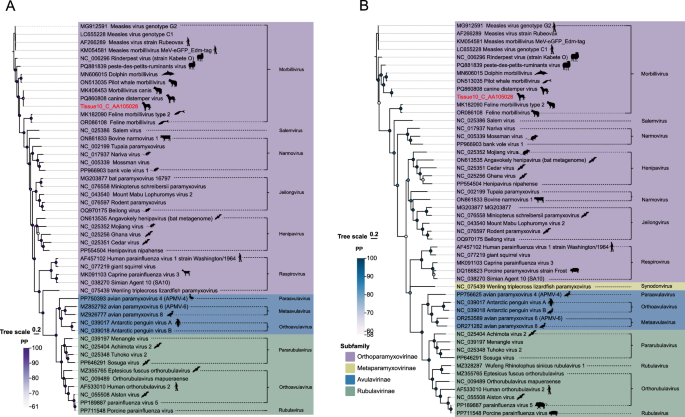
Phylogeny of Paramyxoviridae based on L and F proteins. A L protein tree. B F protein tree. Sequences from this study are marked in red. Subfamily and genus annotations are shown in the legend
Polyomaviridae
Polyomaviridae is a family of small double-stranded DNA viruses that widely infect vertebrates. Their genetic material consists of circular double-stranded DNA, approximately 5 kbp in length [46]. According to the International Committee on Taxonomy of Viruses (ICTV), the Polyomaviridae comprises four genera: Alphapolyomavirus, Betapolyomavirus, Gammapolyomavirus, and Deltapolyomavirus. While Gammapolyomavirus primarily infects birds, the other genera can infect mammals and humans [47]. In this study, a complete circular genome of 5,107 bp was assembled de novo. Phylogenetic analysis based on the Large T antigen (LT) protein revealed that the target sequence shares the highest similarity (97%) with a canine polyomavirus (GenBank: NC_034456) [48] (Fig. 4). The sequence NC_034456 belongs to Betapolyomavirus and was identified in respiratory swab samples collected from canines in the United States.
Fig. 4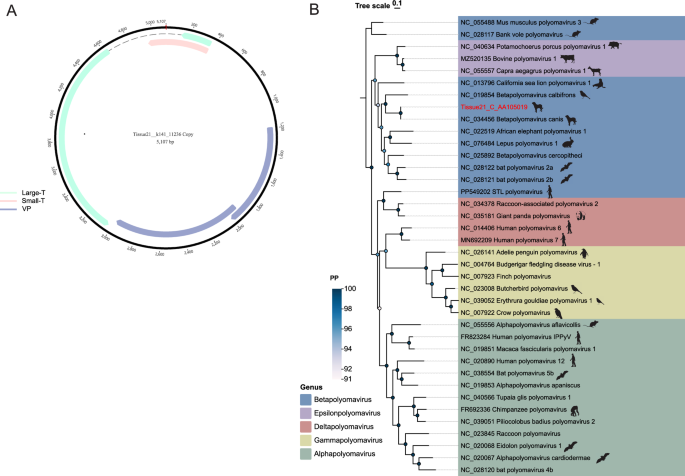
Polyomaviridae genomic structure and phylogeny. A Schematic of complete polyomavirus genome assembled in this study. B LT protein phylogenetic tree. Sequences from this study are highlighted in red. Scale bar indicates evolutionary distance
Parvoviridae
Parvoviridae comprises viruses with a linear single-stranded DNA (ssDNA) genome of approximately 4–6 kb in length. The genome primarily consists of two open reading frames (ORFs), which encode the non-structural protein 1 (NS1) and the viral capsid protein (VP) [49]. Currently, parvoviruses are known to infect vertebrates and insects, and Parvoviridae is classified into three subfamilies: Parvovirinae, Densovirinae, and Hamaparvovirinae [50, 51]. Parvovirus infections are associated with various diseases; for example, Canine parvovirus (CPV) is a novel virus that emerged in the late 1970s, capable of infecting canines and primarily causing clinical gastroenteritis in puppies. Over time, viral variants have gradually emerged, with one such variant designated as CPV-2a. The CPV-2 variant has remained prevalent among wild carnivores and has been well-documented in multiple countries worldwide, whereas human parvovirus B19 infection can result in aplastic anemia, particularly in immunocompromised individuals [52,53,54].
In this research, seven complete Parvoviridae genomes containing the full NS1 coding region were successfully assembled. Comparative analyses revealed that four viral sequences were widely distributed across multiple sample libraries (Fig. 5A). The sequence represented by Tissue24_C_AA105026 was detected in 22 out of the 24 sample libraries analyzed in this study, demonstrating high prevalence among the canine specimens examined. Furthermore, the sequence represented by Tissue3_C_AA105020 was identified in 10 sample libraries (Fig. 5B). These findings suggest that the aforementioned viruses may have important epidemiological implications in canine hosts, warranting further investigation into their transmission mechanisms and potential pathogenicity.
Fig. 5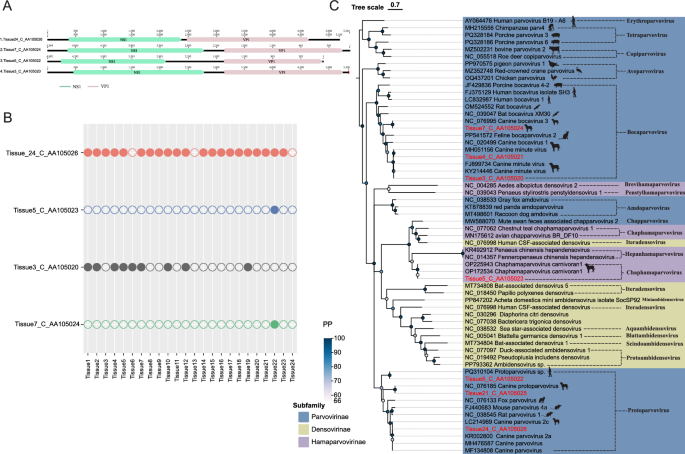
Parvoviridae genome organization and phylogeny. A Genomic structures of four parvoviruses. B Prevalence of these parvoviruses across 24 libraries. C NS1 protein phylogenetic tree. Sequences identified in this study are shown in red
Phylogenetic analysis based on the NS1 protein revealed that the target sequences clustered with members of the Parvovirinae and Hamaparvovirinae (Fig. 5C). Notably, five genera within the Parvovirinae—Erythroparvovirus, Tetraparvovirus, Bocaparvovirus, Protoparvovirus,and Dependoparvovirus—have been reported to include human-infecting viruses [49]. Among the identified sequences, Tissue7_Parvovirus, Tissue4_Parvovirus, and Tissue3_C_AA105020 exhibited over 99% sequence homology with known canine parvovirus sequences (GenBank: NC_076995, MH051156, FJ899734) and were classified within the Bocaparvovirus. The phylogenetic analysis revealed that Tissue24_C_AA105026, Tissue21_C_AA105025, and Tissue5_C_AA105022 clustered within the same clade as members of Parvovirinae, and were classified within the Protoparvovirus. Among these, Tissue24_C_AA105026 and Tissue21_C_AA105025 exhibited the highest sequence homology with canine parvovirus strains (GenBank: KR002800 and NC_076185), showing 99.70% and 99.40% identity respectively, this suggests that the target sequence is widely prevalent in canines. Notably, Tissue5_C_AA105022 demonstrated 100% sequence identity with the PQ310104 reference strain. NCBI records indicate that sequence PQ310104, classified within Protoparvovirus, was detected in an oral swab sample from a human in Shanghai. Given that parvoviruses sharing ≥ 85% amino acid sequence identity in their NS1 proteins are considered members of the same species, these results suggest that Tissue5_C_AA105022 detected in canine lymph nodes belongs to the same species as the virus detected in human oral specimens (GenBank: OP172534) [55].
Circoviridae
Circoviridae possess a circular single-stranded DNA (ssDNA) genome, and all known circoviruses encode at least two proteins: the capsid protein (Cap) and the replication-associated protein (Rep), both of which are encoded in an ambisense orientation [56]. Circoviridae are widely distributed in nature, primarily infecting birds and mammals, and have significant implications for human health [57, 58]. Previous studies have detected Circoviridae in cerebrospinal fluid samples from individuals with acute central nervous system diseases [59]. Canine circovirus belongs to Circoviridae, it was first described in 2011 and has since been detected in various countries and it has been found in co-infections with other viral pathogens in most cases (e.g., CPV) [60, 61]. In addition, CaCV has also been reported in clinically healthy dogs, indicating that its presence is not always associated with overt disease [62].
In this study, six complete circular genome sequences were successfully assembled. Phylogenetic analysis based on the Rep protein revealed that Tissue7_C_AA105031 clustered within the same evolutionary clade as a sequence derived from pigeons (GenBank accession: MW181975) with 100% sequence homology, suggesting potential avian-to-canine cross-species sharing (Fig. 6). Furthermore, Tissue1_C_AA105029, Tissue4_C_AA105030, and Tissue23_C_AA105033 clustered with a canine-derived sequence (GenBank accession: PP691203), exhibiting sequence homologies of 97.69%, 97.69%, and 97.36% respectively, indicating they belong to the same species. All these sequences grouped with members of Circovirus and can thus be classified as circoviruses.
Fig. 6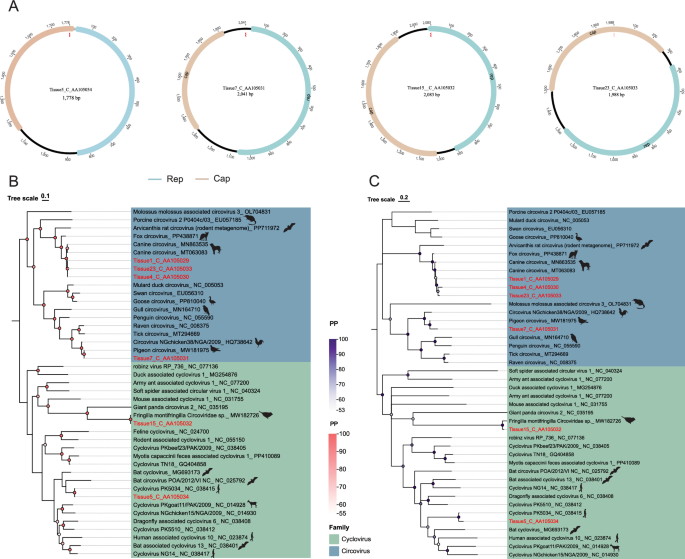
Circoviridae genome organization and Bayesian phylogeny. A Genomic structures of four circoviruses. B.C Bayesian trees based on Rep (left) and Cap (right) proteins. Sequences from this study are highlighted in red
Notably, Tissue15_C_AA105032 clustered with a sequence derived from a tit (GenBank accession: MW182726) within the unclassified circovirus group, exhibiting 100% sequence identity, which suggests they represent the same viral species. However, we cannot ascertain whether this virus detected in canine lymph nodes originated from active infection rather than passive carriage through ingestion of avian tissues. Moreover, Tissue5_C_AA105034 exhibited 99% sequence homology with the human-derived sequence NC_038415. NC_038415 belongs to human-associated cycloviruses, which were first identified in 2010 through high-throughput sequencing of fecal and cerebrospinal fluid samples from human populations in Southeast Asia (Vietnam, Cambodia), including strains such as Cyclovirus VN and Cyclovirus PK [63, 64]. While this sequence shows high similarity to human-associated strains, we cannot determine whether it represents active infection in dogs or reflects passive carriage.
In the present study, we identified viral sequences in canine lymph node tissue showing 99% homology with human-associated cycloviruses. A preliminary hypothesis of potential cross-species transmission could be postulated. In addition, we found topological differences between the Cp-based and Rep-based phylogenetic trees, which may be attributed to genetic recombination during viral evolution.
Genomoviridae
Genomoviridae is a family of single-stranded circular DNA viruses with a genome size of approximately 2–2.4 kb, exhibiting high genetic diversity [65]. According to the current species classification criteria, viruses with less than 78% whole-genome sequence identity are classified as new species [66]. In this research, we identified a complete Genomoviridae genome with a total length of 2,156 bp. This genome contains two major open reading frames (ORFs), encoding the Rep and Cp proteins, which are transcribed in opposite directions. Additionally, we observed the presence of an intron structure within the gene encoding the Rep protein. BLASTn analysis against the NCBI database revealed that the sequence with the highest similarity to our target sequence was derived from an air sample (GenBank: MW678893) with an identity of approximately 77%, indicating that our identified sequence represents a novel species. A phylogenetic tree constructed based on the whole-genome showed that our sequence clustered with MW678893, belonging to unclassified Genomoviridae (Fig. 7).
Fig. 7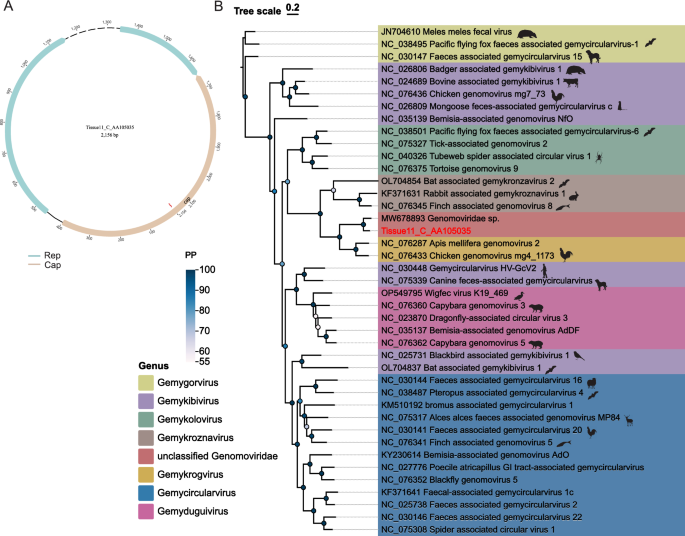
Genomoviridae genomic structure and phylogeny. A Genomic diagram of the novel virus identified in this study. B Phylogenetic tree based on the complete genome; sequences from this study are highlighted in red. Scale bar indicates evolutionary distance