
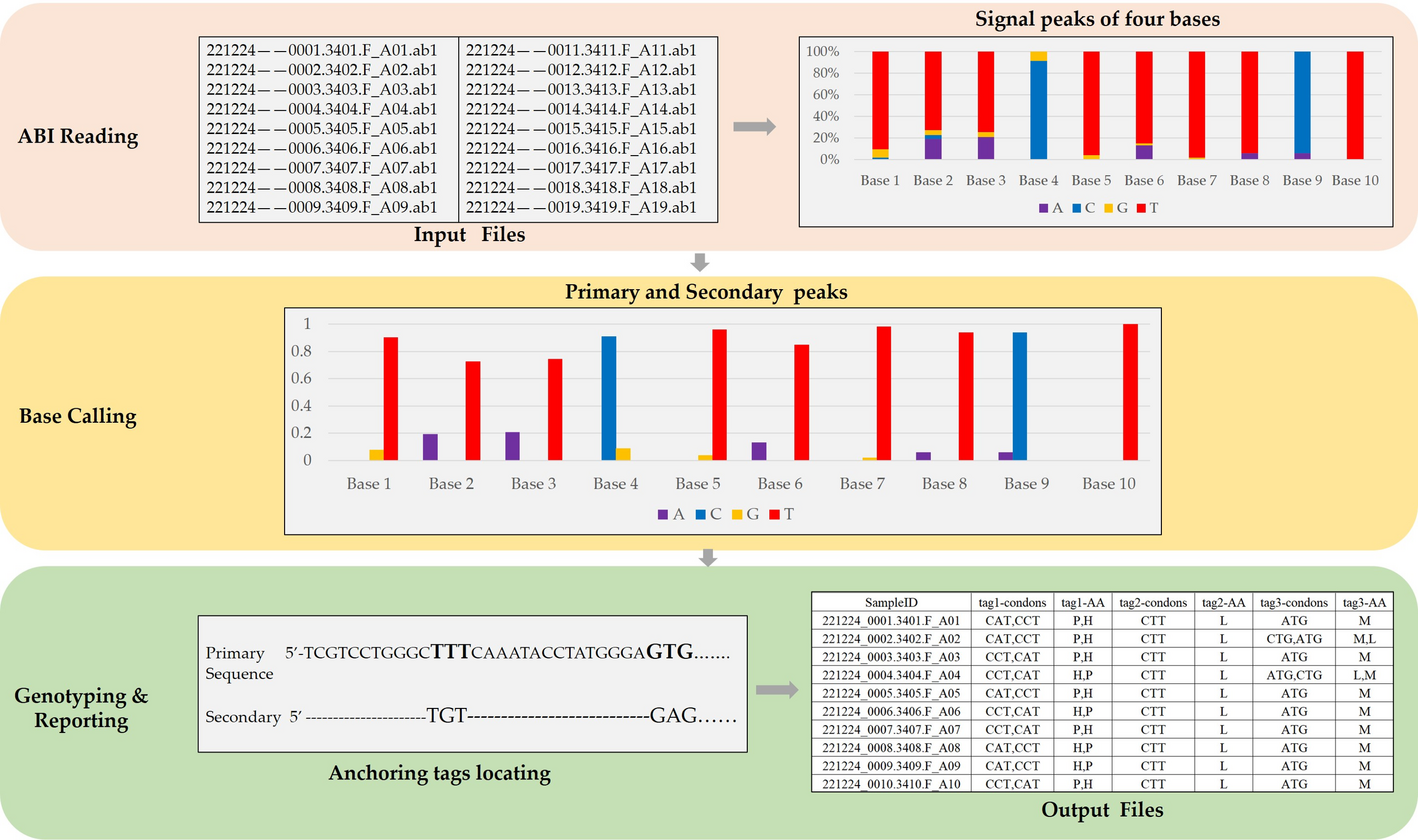
Software architecture
SAGPEK mainly processes Sanger sequencing results of two kinds of samples: diploids such as humans and livestock animals and haploids such as viruses and bacteria. For diploids, SAGPEK reports the homologous or heterozygous genotypes of targeted loci. However, for haploids, it reports haplotypes. If the detected loci are located in the coding region, SAGPEK also reports alterations in amino acids.
SAGPEK performs three main functions (see Fig. 1). The first is reading in the ABI-format files as inputs. It extracts normalized fluorescence intensity signals for A, C, G, T, detects positions of maximum peaks in each channel and calculate corresponding peak amplitudes. Second, SAGPEK performs base calling to determine the primary and secondary bases by identifying the highest peak intensity and detecting if a secondary signal reaching ≥ 33% of the strongest peak. Third, SAGPEK maps anchoring sequences to primary bases to locate the targeted loci, obtains their genotypes and outputs results to a result file. Users should feed the SAGPEK anchoring sequence for each target locus, which is usually a short sequence between 6 and 10 nucleotides just in front of the target locus from the orientation of the 5′ primer. SAGPEK searches anchoring sequences in the primary sequence and locates the target locus. After the coordinates of the target locus in the primary sequence are obtained, SAGPEK can judge whether the locus is homozygous or heterozygous and determine the genotype. In some situations, Sanger sequencing is performed from the reverse orientation, e.g., from 3′ to 5′, in which case the − r option should be invoked, and the anchoring sequences should be in the 5′ to 3′ orientation. SAGPEK can also report amino acid alterations for each locus for each input ABI file. By default, amino acid alterations are generated in the output files. If no amino acid alteration needs to be generated, users should turn off the function with the -AA option (by default, this function is on). If enabled, the amino acid alteration option will also process loci in non-coding regions, returning “NA” for such sites. This does not affect genotype calling but may add unnecessary “NA” entries, so disabling it is recommended for non-coding analyses.
Fig. 1
Schematic of SAGPEK implementation. The input files are in ABI format and are read in SAGPEK to generate intermediate files that record the signal intensities of four bases (A, C, G, T) for each base position of each input file. Then, SAGPEK performs base calling at each base position and records the status flag indicating whether the site is heterozygous or homozygous. At this step, SAGPEK generates two sequences: the primary sequence, which consists of bases with the highest signal intensity, and the secondary sequence, which consists of bases with the second highest signal intensity. Next, SAGPEK locates target loci by mapping anchoring sequences to the primary sequence. The anchoring sequences are either built in the SAGPEK or provided by users. After successfully mapping the anchoring sequences, SAGPEK obtains the coordinates of the target loci. SAGPEK extracts the primary and secondary bases, and the status flag indicates homozygosity or heterozygosity. By judging the status flag, SAGPEK determines the target loci genotypes and writes the results to the output files. SAGPEK also reports the alteration of corresponding amino acids to the output files
Software functionalities
SAGPEK has built-in anchoring sequences for common drug-resistant loci of HBV, the loci of inherited metabolic diseases of the newborn population, mutations in citrullinemia (CN) [38] and deficiency of uridine monophosphate synthase (DUMPS) [28] in cattle. For each locus, we provide its anchoring sequence in Supplementary Table S1, enabling users to identify and select appropriate anchors for their specific workflows.
Users need to use the “-type HBV” option to invoke automapping of targeted loci for common drug-resistant loci of HBV. SAGPEK stores anchoring sequence tags for 25 common drug-resistant mutations in the reverse transcriptase domain (POL/RT) of the HBV polymerase (Fig. 2). The polymerase number is defined according to the serial number of amino acids of the reverse transcriptase on the basis of the HBVdb X20763 protein number (https://hbvdb.lyon.inserm.fr/HBVdb/). SAGPEK reads in the input ABI files, generates nucleotide sequences, and locates the drug-resistant mutation loci by mapping anchoring tags. If the anchoring tags are mapped to the nucleotide sequences, the sequence of the corresponding codon is determined and translated to the amino acid. By comparing the amino acid sequence with the built-in amino acid sequence reference, SAGPEK reveals whether the mutation occurs and what the mutation is. If one of the 25 common loci is not detected, SAGPEK will generate the unavailable field in the output file for the locus.
Fig. 2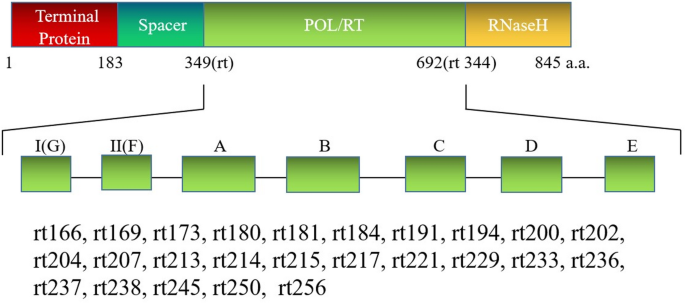
Schematic diagram of the detection of HBV polymerase gene mutations in SAGPEK. SAGPEK stores 25 anchoring sequences corresponding to 25 common drug-resistance mutations ranging from rt166 to rt256. All these mutations are located within the POL/RT domain of the HBV polymerase. The POL/RT domain is numbered from rt1 (first amino acid) to rt344 (last amino acid) according to standard HBV polymerase nomenclature
For inherited metabolic diseases of the newborn population, SAGPEK stores anchoring sequence tags for the 14 common mutation sites of the phenylalanine hydroxylase (PAH) gene (Supplementary Table S1, Fig. 3), which is the critical gene in phenylketonuria (PKU) disease [39,40,41]. Phenylketonuria is an autosomal recessive genetic disease. Owing to gene mutation, phenylalanine cannot be metabolized generally in the body, and abnormal accumulation of harmful metabolites produced by its bypass metabolism leads to severe mental retardation and neurobehavioral abnormalities. Pathogenic mutations, including missense mutation, splicing site mutation, silent mutation, and nonsense mutation, have been detected in 13 exons (exon, E), introns (I), the 5′ untranslated region (5′-UTR), and the 3′ untranslated region (3′-UTR) of the PAH gene. The mutation numbering is based on naming rules according to the BIOPKU database (http://biopku.org/home/home.asp).
Fig. 3
Schematic diagram of the human PAH gene structure. The PAH gene has 13 coding regions and consists of 452 amino acids. SAGPEK stores 14 anchoring sequences for 14 common mutations in the PAH gene ranging from p.48 to p.415, where p.48 represents the 48th amino acid and p.415 represents the 415th amino acid of the whole protein. The 14 mutations are dispersed in the three domains of the PAH protein
For cattle genetic defects, SAGPEK stores anchoring sequences for mutations in citrullinemia (CN) [38] and deficiency of uridine monophosphate synthase (DUMPS) [28]. For CN, the 86th amino acid codon encoded by the argininosuccinate synthetase (ASS) gene on chromosome 11 of dairy cows has a nonsense mutation (CGA->TGA), resulting in the loss of the synthetic peptide chain, which results in the loss of ASS function in recessive homozygous individuals. For DUMPS, there is a C/T point mutation at the 405th codon of the C-terminus encoded by the UMPS gene, which leads to the mutation of the CGA of the arginine codon into the TGA terminator and results in the loss of 76 amino acids in uridine synthase.
In addition to the built-in anchoring sequence tags, SAGPEK takes user-defined anchoring sequence tags with the “-type custom” option. Users should provide a header-free file with two columns: the first lists the tag names, and the second lists the anchoring sequences. If amino acid alterations need to be generated in the output file, ensure that the anchoring sequences are located just in front of the first base of the investigated codons.
While SAGPEK’s primary function is to generate genotype files, it also supports the optional output of sequencing chromatograms. By specifying the parameter -chromatogram on, users can generate chromatogram images for selected samples, enabling visual inspection of sequencing peaks.
Illustrative examples
SAGPEK is implemented with the Perl language. Users should install an Perl interpreter before using SAGPEK. SAGPEK can work in both Windows and Unix-like platforms. After installing the dependencies, download and unzip SAGPEK from https://github.com/JINPENG-WANG/SAGPEK. A folder with five subfolders (ABI, chromatogram, Custom, Genotype and lib) and one executive file (SAGPEK.pl) will be generated. The SAGPEK.pl script is used to execute the program. The ABI-format input files should be placed in the ABI folder. SAGPEK is easy to use. We describe its usage below. A summary of options is provided in Supplementary Table S2.
Before using, users should install an Perl interpreter, such as from the ActiveState website (https://www.activestate.com/products/perl/). After the installation, the directories in which executable programs are located should be added to the PATH environment variable. Before running the package, users should put the ABI-format files generated by Sanger sequencing technology into the “ABI” file folder of the SAGPEK package. Then, the command prompt window is opened, and the working directory is changed to the SAGPEK package directory with the “cd” command. The program is run with the “perl SAGPEK.pl -type [TYPE]” command, where the TYPE should be replaced with HBV, PAH, cattle_CN, cattle_DUMPS, TEST, or custom according to the actual situation. If the “custom” type is used, users should also provide the name of a file storing the customized anchoring sequences located in the “Custom” directory and add the file name to the command as “perl SAGPEK.pl -type custom -tag ab.tags.txt”. The tag file must consists of two columns. The first column stores the names of the anchoring sequences, and the second column stores the sequences. After completion, a result file consisting of genotypes or amino acid alterations is generated in the Genotype Directory. The explanation of the output files describing each output column in detail including name, definition and example values is listed in Supplementary Table S3. Users can also generate chromatograms for each input ABI file with the “perl SAGPEK.pl -type TEST -chromatogram on” command.
Usage examples
We list a set of common uses of SAGPEK below. We provide several ABI format files in the ABI folder as test dataset, which can be used to test SAGPEK.
The SAGPEK can be used in Windows/Linux/Mac systems as follows:
$ perl SAGPEK.pl -type TEST
This will generate a test output file (TEST.genotype.txt) in the ./SAGPEK/Genotype/ directory, allowing users to verify correct installation and execution.
If the -AA is set to off, the SAGPEK can be run as follows:
$ perl SAGPEK.pl -type TEST -AA off
This will generate a test output file which only reports the genotypes of tested loci but not the amino acids alterations.
Users can also test SAGPEK with the custom type. We have provided an tag file named “ab.tags.txt” in the Custom folder which stores anchoring sequences and their names. Users can run SAGPEK with the following command:
$ perl SAGPEK.pl -type custom -tag ab.tags.txt
This will generate an output file (Custom.mutants.txt) in the ./SAGPEK/Genotype/ directory, which stores genotypes of loci mapped with the anchoring sequencings in the “ab.tags.txt” file.
For the HBV, PAH, cattle_CN, and cattle_DUMPS types, SAGPEK has built-in anchoring sequences.
If chromatogram is needed, the SAGPEK can be run with the following command:
$ perl SAGPEK.pl -type custom -tag ab.tags.txt -chromatogram on
This will generate the chromatogram plots in the chromatogram directory for all investigated input ABI files.