
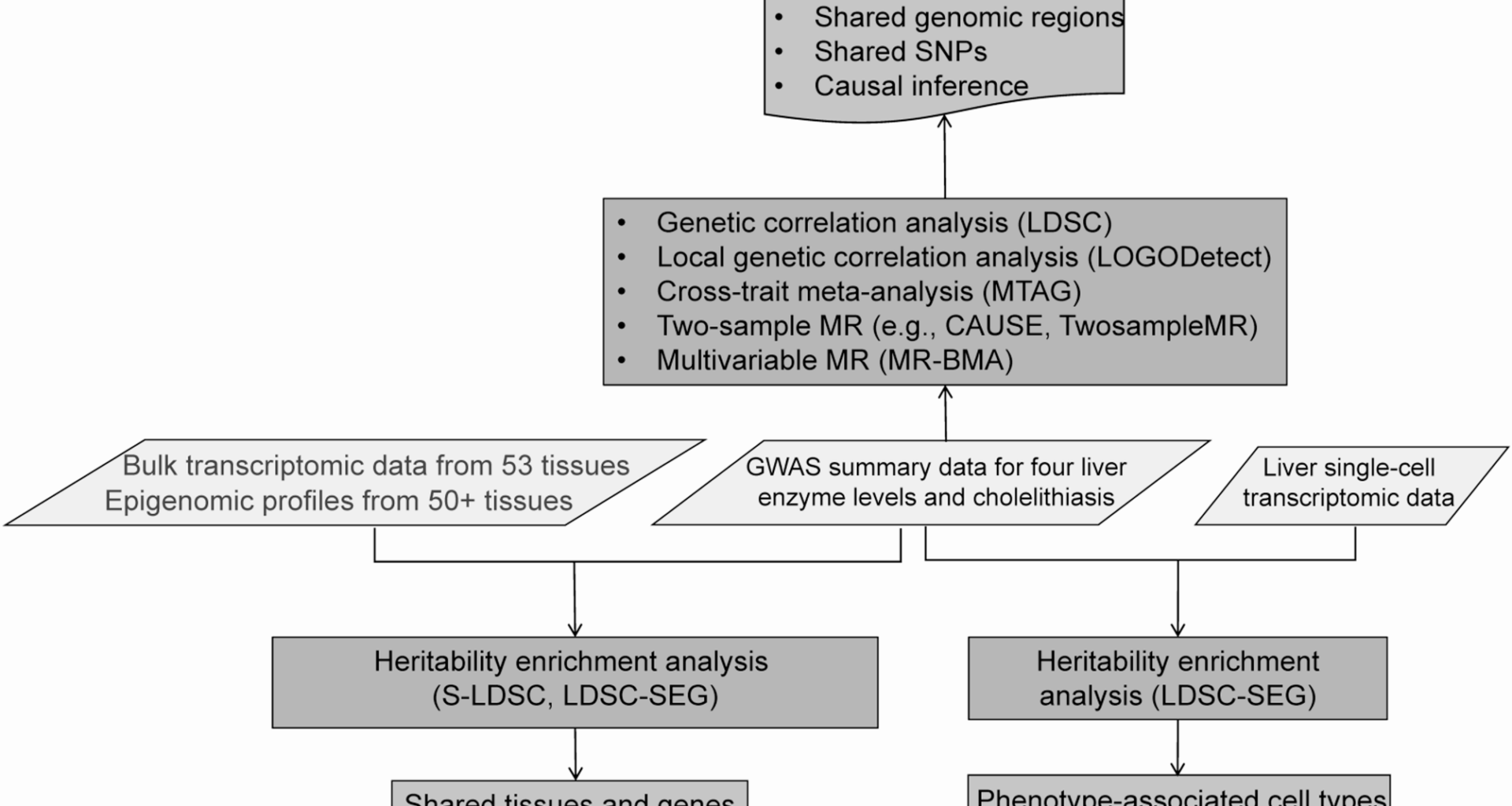
GWAS summary data
Genome-wide association study (GWAS) summary statistics were obtained from three publicly available databases: OPEN GWAS [11], GWAS Catalog [12], and the FinnGen project [13]. Data from OPEN GWAS and GWAS Catalog were directly downloadable. Access to FinnGen project data required completion of an online form on the official website, with only the name, institutional affiliation, country, and email address needed. Upon submission, an email containing download links to the full set of GWAS summary statistics was received. The public sources and download links for all datasets used in this study are summarized in Table S1.
Liver enzymes:
Pazoki et al. performed a two-stage GWAS of serum levels for ALT, ALP, and GGT [14]. The discovery phase comprised 437,438 UK Biobank participants of European ancestry (aged 40–69) recruited during 2006–2010. Exclusion criteria covered withdrawn consent, uncertain pregnancy status, sex mismatches, poor genotyping quality, and hepatobiliary disorders. Liver enzyme measurements used baseline blood samples processed at − 80 °C with subsequent standardized assays. Final discovery sample sizes were 437,267 (ALT), 437,438 (ALP), and 437,194 (GGT). The replication phase (N = 315,572) confirmed 75% of initial loci. For our study, we utilized discovery-phase summary statistics from GWAS Catalog (accessions: GCST90013405-ALT, GCST90013406-ALP, GCST90013407-GGT).
Additionally, we used GWAS summary data on ALP and AST from the Neale Lab’s UK Biobank analysis, which involved 361,194 European participants. These measurements were also based on single-time-point samples collected at baseline, and the data are available from the OPEN GWAS database [11] under accession codes ukb-d-30650_raw and ukb-d-30610_raw. However, ALP data from the Neale Lab was only utilized for two-sample Mendelian randomization (2SMR) due to heterogeneity in SNPs compared to the Pazoki dataset.
Cholelithiasis:
GWAS summary statistics for cholelithiasis were obtained from the FinnGen project (release finngen_R6_K11_CHOLELITH) [13]. The cohort comprised 23,084 unrelated Finnish adults of European ancestry (16,086 females; 6,998 males) and 231,644 controls recruited through nationwide biobank networks with linked electronic health records. Overall cholelithiasis prevalence was 8.88% (females: 10.95%; males: 6.19%), with mean age at first diagnosis of 51.2 years (females: 48.3 years; males: 57.7 years). Cases were defined by ICD-10 code K80 without stone-type differentiation. Genotyping quality control and association analyses implemented SAIGE with adjustment for sex, age, ten principal components, genotyping batch, and relatedness. Crucially, the liver enzyme (UK Biobank) and cholelithiasis (FinnGen) GWAS datasets represented independent cohorts with no sample overlap, thereby minimizing bias in our 2SMR.
Others:
For multivariable Mendelian randomization, we also used GWAS summary data for low-density lipoprotein cholesterol (LDL-C) [15], body mass index (BMI), hip circumference, and total cholesterol (TC) from European populations. The LDL-C data, obtained from a study by Tom G. Richardson et al., included 440,546 participants of European ancestry [15]. BMI, hip circumference, and TC data were sourced from the OPEN GWAS database [11] (identifiers: ukb-a-248, ukb-a-388, met-d-Total_C) with sample sizes of 336,601 for BMI and hip circumference, and 115,078 for TC.
Bulk RNA transcriptome data
We utilized bulk RNA sequencing data from the Genotype-Tissue Expression (GTEx) project for stratified heritability enrichment analysis. Specifically, gene expression data from GTEx V6 (comprising 544 healthy donors) were used, following the approach described by Finucane et al. [16], to evaluate tissue-specific enrichment across 53 human tissues. For Summary-data-based Mendelian Randomization (SMR) analysis, we employed cis-expression quantitative trait locus (cis-eQTL) summary statistics from GTEx V7, which were derived from 153 liver tissue samples. These data enabled the investigation of gene-level associations between gene expression and traits.
Chromatin accessibility and histone modification data
Chromatin accessibility and histone modification profiling data were obtained from the Roadmap Epigenomics Consortium and the ENCODE-Tissue Expression (EN-TEx) resource [17, 18]. The dataset comprises six epigenetic features: DNase I hypersensitive sites (which indicate open chromatin regions) and five histone modifications—H3K27ac, H3K4me3, H3K4me1, H3K9ac, and H3K36me3—that are commonly associated with promoters, enhancers, and transcriptional regulation. These features were profiled across 88 major cell types or tissue subgroups. Additional details regarding data processing can be found in the original study by Finucane et al. [19].
Single-cell transcriptome data
To assess stratified heritability enrichment at the cellular level, we initially utilized gene expression data from seven distinct mouse liver immune cell types sourced from ImmGen Microarray Phase 1 [20] and ImmGen Microarray Phase 2 [21]. Details of the data processing procedures are available in the original study by Finucane et al. [19]. Additionally, we incorporated a gene expression matrix derived from single-cell transcriptomic data of human liver cirrhosis, processed by Xiangyu Ye et al. [22]. These data were obtained from the Gene Expression Omnibus (GEO) database [23]. Single-cell data from five cases of liver cirrhosis in dataset GSE136103 were processed by Xiangyu Ye et al. [22], resulting in the identification of 16 cell types.
Estimating genetic correlation using LD score regression
Linkage Disequilibrium Score Regression (LDSC) is a widely used statistical method in GWAS for calculating linkage disequilibrium (LD) scores at each SNP locus, thereby enabling the estimation of trait heritability and genetic correlation between traits [24, 25]. In this study, we first evaluated the heritability of five traits. We then used LDSC (v1.0.1) to estimate the genetic correlation between liver enzymes levels and cholelithiasis, aiming to identify which enzyme shows a stronger genetic association with cholelithiasis. This was done using an intercept restriction, and a sensitivity analysis was performed with the unconstrained LDSC model. Assuming no population stratification and with available information on sample overlap and phenotypic correlations, the intercept can be set to a specific value, which reduces the standard error by approximately 30%. When sample overlap is absent, the intercept is set to zero, a method referred to as constrained intercept LDSC [24, 25]. In all LDSC analyses, we applied the baseline model, which incorporates a comprehensive set of functional genomic annotations to enhance the accuracy of heritability and genetic correlation estimates. Statistical significance was defined as P < 0.05 without multiple testing correction, due to the limited number of pairwise comparisons.
Estimating local genetic correlation
Local genetic correlation quantifies the shared genetic influences between two traits within a specific genomic region (e.g., a chromosome segment), distinct from genome-wide genetic correlation. While genome-wide genetic correlation provides valuable insights into the shared genetic architecture of traits, it may overlook localized genetic effects and pleiotropic associations. To address this limitation, we conducted a local genetic correlation analysis using the LOcal Genetic cOrrelation Dectector (LOGODetect, v1.0.0) [26]. LOGODetect is a pioneering method that employs scan statistics to identify genomic regions exhibiting local genetic correlation between two complex traits. Unlike other approaches, LOGODetect does not rely on pre-specified genomic regions but instead automatically detects regions of significant local genetic correlation, offering higher resolution and greater statistical power [26]. In this study, we analyzed GWAS summary statistics for multiple liver enzymes (ALT, GGT, ALP, and AST) and cholelithiasis using LOGODetect. Analyses incorporated LD reference data from individuals of European ancestry in the 1000 Genomes Project. The genome was partitioned into blocks of up to 5,000 SNPs, with a scanning interval set at 50. LOGODetect outputs Bonferroni-adjusted P-values by default, and statistical significance was defined as adjusted P < 0.05.
Cross-trait meta-analysis
To further investigate shared genetic loci between liver enzyme levels and cholelithiasis, we performed a meta-analysis using the Multivariate Trait Analysis of GWAS (MTAG, v1.0.8) [27]. This method increases statistical power by effectively enlarging the sample size, thereby facilitating the identification of novel loci jointly associated with liver enzymes and cholelithiasis. MTAG assumes equal heritability and perfect genetic covariance between traits. Cross-trait meta-analyses were conducted for each liver enzyme – cholelithiasis pair.
To address potential inflation of false positives introduced by multi-trait modeling, we applied the –fdr flag to estimate the upper bound of the max false discovery rate (maxFDR). This conservative measure reflects the maximum expected proportion of false positives among genome-wide significant findings, thereby improving the interpretability and robustness of the results. Given that the GWAS summary statistics for liver enzymes and cholelithiasis were derived from non-overlapping samples, the -no_overlap option was also used to satisfy MTAG’s assumption of sample independence. All other parameters were set to their default values. Genotype data from individuals of European ancestry in the 1000 Genomes Project Phase 3 were used as the LD reference panel. Genome-wide significant loci (P ≤ 5e-8) were identified using PLINK v1.9, with an LD threshold of R² < 0.001 and a physical distance cutoff of 10,000 base pairs. Only loci that reached genome-wide significance within the reference panel were retained. Loci uniquely identified through MTAG were considered newly discovered shared loci.
Association statistics generated by MTAG were adjusted using the LD Score Regression intercept, which accounts for confounding factors such as population stratification, cryptic relatedness, and sample overlap. Therefore, the P-values reported by MTAG were already corrected and were used directly to assess statistical significance.
Two-sample Mendelian randomization
We first conducted 2SMR analyses using the TwoSampleMR package (v0.5.6), applying five established methods: inverse variance weighted (IVW), MR-Egger, weighted median, weighted mode, and simple mode. Among these, IVW was used as the primary method due to its higher statistical power under the assumption that all instrumental variables are valid or that any pleiotropy is balanced. The additional methods were used for sensitivity analyses to assess the robustness of the results under different pleiotropy assumptions, with MR-Egger allowing for directional pleiotropy and the weighted median providing valid estimates even when up to 50% of instruments are invalid.
The 2SMR analyses adhered to three core assumptions [28]: (1) SNPs are strongly associated with the exposure (relevance); (2) they are independent of confounders (independence); (3) they influence the outcome only through the exposure (exclusion restriction). To ensure relevance, SNPs were selected based on genome-wide significance (P ≤ 5 × 10⁻⁸) and LD pruning (R² < 0.001, 10,000 kb), using the 1000 Genomes Project (European ancestry) as reference. F statistics were calculated using formula \({\rm{F}} = \frac{{{\rm{N}} – {\rm{k}}-1}}{{\rm{k}}} \times \frac{{{{\rm{R}}^2}}}{{1 – {{\rm{R}}^2}}}\) to assess instrument strength and reduce the risk of weak instrument bias. To address independence and exclusion restriction, pleiotropic SNPs were identified using the Phenoscanner database [29]. Mendelian Randomization Pleiotropy RESidual Sum and Outlier (MR-PRESSO, v1.0) [30] was used to detect and remove variants likely introducing horizontal pleiotropy. To reduce heterogeneity, which may signal invalid instruments, we applied RadialMR (v1.0) [31] to identify and exclude outlier SNPs. Causal estimates were obtained using the IVW method. Sensitivity analyses included the MR-Egger intercept (pleiotropy), Cochran’s Q statistic (heterogeneity), and leave-one-out analysis (influential SNPs) to ensure robustness.
Given that traditional MR approaches may include SNPs violating instrumental variable assumptions—thereby introducing pleiotropy and inflating false-positive rates—we conducted an additional causal inference using CAUSE (v1.2.0.0335) [32]. Compared to conventional MR approaches that rely solely on genome-wide significant SNPs, CAUSE improves statistical power and reduces false positives by leveraging genome-wide data. It also accounts for potential sample overlap between exposure and outcome datasets, enabling analysis of the full available sample size [32]. Causal inferences were based on a nominal significance threshold of P < 0.05, without formal correction for multiple testing due to the limited number of trait pairs examined.
Multivariable mendelian randomization
Due to the inability of CAUSE and IVW methods to address collinearity between ALT and GGT, we applied Mendelian Randomization Bayesian Model Averaging (MR-BMA, v0.1.0) [33] to validate the findings from the 2SMR analysis. This method is specifically designed to disentangle exposure variables with high genetic correlation by evaluating all possible combinations of exposures through weighted regression models. Multivariable MR extends the conventional two-sample MR approach by relaxing the assumption that each genetic instrument is exclusively associated with a single exposure. Instead, it allows instruments to be associated with multiple exposures, while still requiring that the core assumptions of instrument relevance, exclusivity, and exchangeability are satisfied [33]. We extracted genome-wide significant SNPs and applied LD clumping to ensure instrument independence. SNPs that were not available in the cholelithiasis dataset or that were associated with known confounders were excluded to preserve the assumption of exclusivity. Six exposures—ALT, GGT, BMI, LDL, TC, and hip circumference—were included in the MR-BMA model. Weighted regression was performed across all combinations of these traits.
Stratified heritability enrichment analysis
Stratified heritability enrichment evaluates whether particular genomic annotations—such as liver-specific regulatory elements—contribute disproportionately to trait heritability, thereby facilitating the identification of tissues and cell types associated with the phenotype. To investigate tissue and cell type-specific enrichment in cholelithiasis and serum liver enzyme levels, we utilized LDSC (v1.0.1) methods including specifically expressed genes LDSC (LDSC-SEG) [34] and stratified LDSC (S-LDSC) [19]. While S-LDSC partitions heritability based on chromatin accessibility and histone modification profiles [19], LDSC-SEG extends this framework by incorporating tissue-specific gene expression data for heritability partitioning [34]. We applied LDSC-SEG to evaluate SNP heritability enrichment of ALT, GGT, ALP, AST, and cholelithiasis across various tissues. This analysis followed the methodology described by Finucane et al., using baseline models, standard regression weights, and transcriptomic data from GTEx v6. Additionally, we conducted cell-level enrichment analysis leveraging single-cell RNA sequencing data focused on liver tissue. In both stratified LDSC and LDSC-SEG analyses, the baseline model includes a comprehensive set of functional genomic annotations to control for confounding factors and enhance the robustness of heritability partitioning. Statistical significance was defined as P < 0.05 without correction for multiple testing, given the exploratory nature of the analyses.
Identification of shared effect genes using SMR
We used the SMR [35] method to identify shared genes between liver enzyme levels (ALT, GGT, ALP, AST) and cholelithiasis. SMR integrates summary data from GWAS and eQTL studies to assess potential causal associations between gene expression and phenotypes, using genome-wide significant SNPs as instrumental variables. The observed associations may be influenced by causal effects, pleiotropy, or genetic linkage. Although the HEIDI test is incorporated within SMR to address this issue, distinguishing causal effects from pleiotropy remains challenging [35].
In this study, we integrated tissue-specific eQTL data from GTEx V7, focusing on tissues identified by LDSC-SEG as shared between liver enzymes and cholelithiasis. LD was calculated using a reference panel from the 1000 Genomes Phase 3 Project, and only SNPs with genome-wide significance (P ≤ 5 × 10⁻⁸) were considered. To account for potential heterogeneity, we applied the HEIDI test and used p ≤ 0.05 as the statistical significance threshold, such that SNPs with p-values below this cutoff—indicating significant heterogeneity—were excluded. SMR analyses were performed using default software parameters, including a cis-window size of ± 1 Mb around the transcription start site to enhance biological relevance. Additionally, we applied the SMR method to GWAS summary data generated by MTAG to identify additional shared genes.
Statistical analysis software
The statistical analysis was conducted using various R packages, including “TwoSampleMR”, “CAUSE”, “MR-BMA”, “MR-PRESSO”, and “RadialMR” all of which were implemented on version 4.1.0 of the R statistical software. Various software tools, namely LDSC, LOGODetect, MTAG, and SMR, were employed following their respective tutorials. Detailed information regarding the functions, codes, and execution environments for these R packages and tools can be found at the following web address:
LDSC: https://github.com/bulik/ldsc;
LOGODetect: https://github.com/ghm17/LOGODetect;
MTAG: https://github.com/JonJala/mtag;
PLINK: https://www.cog-genomics.org/plink/1.9;
CAUSE: https://jean997.github.io/cause/index.html;
SMR: https://cnsgenomics.com/software/smr/#Overview;
MR-BMA: https://github.com/verena-zuber/demo_AMD;
MR-PRESSO: https://github.com/rondolab/MR-PRESSO;
TwoSampleMR: https://mrcieu.github.io/TwoSampleMR/index.html;
RadialMR: https://github.com/WSpiller/RadialMR.