
Epidemiological analysis of RVAs
From January 2020 to December 2023, the Shenzhen CDC collected fecal samples from diarrhea patients at four sentinel hospitals, while the Zhuhai CDC collected samples from three sentinel hospitals. A total of 5207 acute gastroenteritis samples were collected, among which 940 cases of viral diarrhea were detected by sentinel hospitals, with 158 of those being positive for rotavirus. The rotavirus positivity rate among AGE patients was 3.03%, and the positivity rate among patients with viral diarrhea was 16.81%. After nucleic acid extraction and PCR testing, we included 57 eligible samples with a CT value less than 28 for sequencing. Among these 57 samples, 48 were from children under 5 years old, accounting for 84.21%, which is consistent with previous studies [33]. The epidemiological information for these 57 samples is detailed (Table S1).
Whole-genome sequencing of 57 rotavirus samples yielded a total of 604 nucleotide sequences, reflecting the segmented nature of rotavirus. Among these, 40 strains were found to possess complete genome sequences, while 13 strains were missing only a single segment. All assembled sequences were successfully submitted to the GenBank database (https://www.ncbi.nlm.nih.gov/genbank/). The related information of those sequences was summarized (Table S2).
Genotype analysis of RVAs
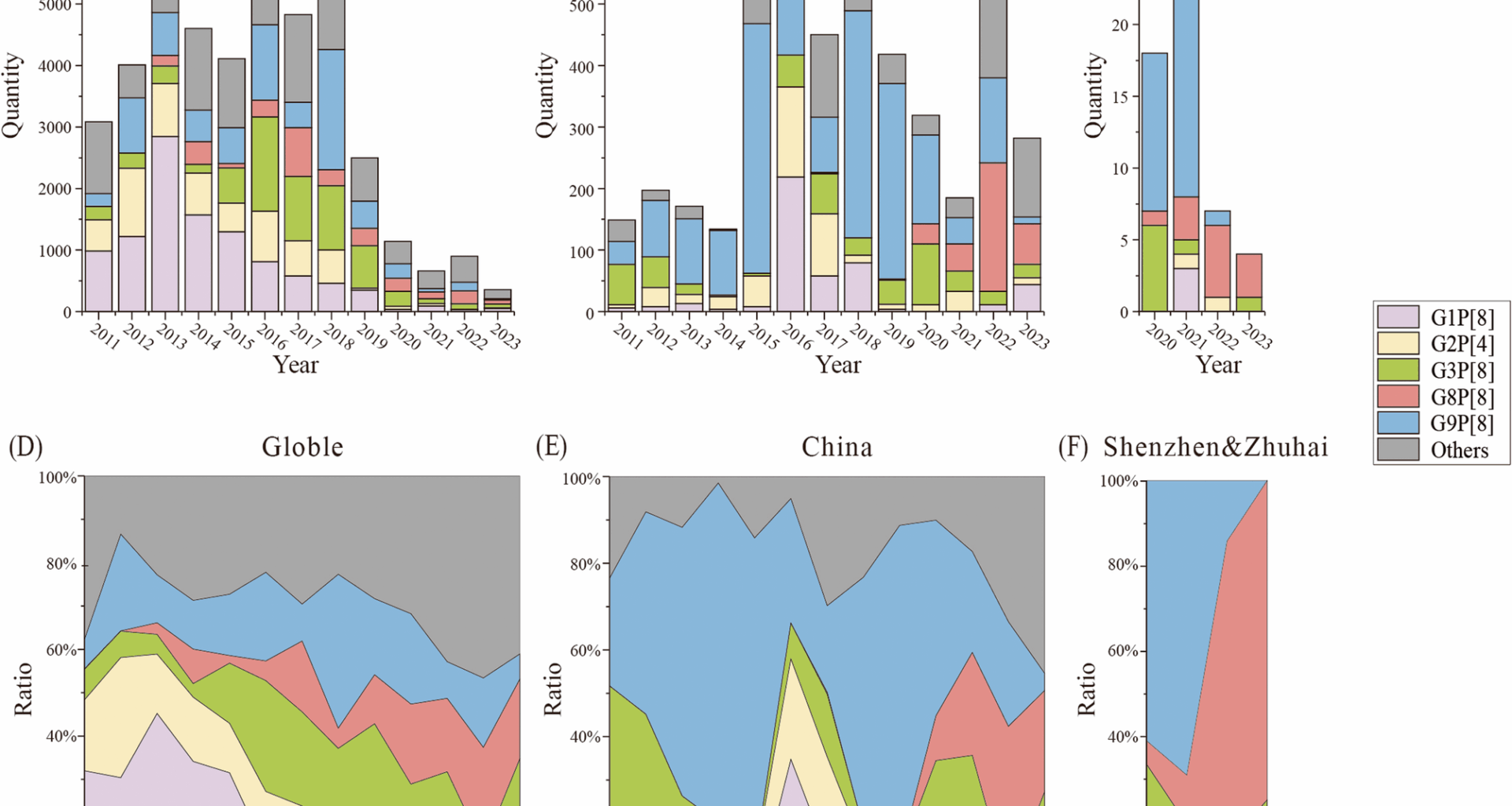
All contributed strains were genotyped successfully (Table S2). Among them, the most common G/P combined genotype was G9P[8] (52.63%, 30/57), followed by G8P[8] (21.05%, 12/57). To investigate the temporal trends of rotavirus genotypes, we compared the evolutionary trends of RVAs at different geographical levels. Specifically, we analyzed the prevalence of rotavirus genotypes post-2011 in both China and across the world, along with the trends observed in the Shenzhen and Zhuhai regions between 2020 and 2023 (Fig. 1).
In terms of genotypes, the distribution patterns demonstrated both similarities and differences across global, national, and regional levels. Globally, the G1P[8] genotype was predominantly circulating before 2015, accounting for the majority of cases (Fig. 1A, D). However, a decline of G1P[8] was observed afterward, with G3P[8] and G9P[8] emerging as the dominant genotypes. G3P[8] exhibited a sharp increase in prevalence, becoming the leading genotype worldwide, whereas G9P[8] increased more gradually. Additionally, G8P[8], though initially rare, had risen to prominence in recent years, even contributing to a decline in dominant strains such as G9P[8]. Similarly in China, the G1P[8] genotype also showed a decreasing trend, albeit with greater fluctuations compared to the global pattern (Fig. 1B, E). In China, G3P[8] and G9P[8] replaced G1P[8] as the predominant genotypes, consistent with global trends. However, a notable divergence occurred after 2020: the G8P[8] genotype, which was rare both globally and in China before this period, experienced a rapid and significant increase, establishing itself as a major genotype in the following years. This trend of G8P[8] expansion aligns with the global rise, but its increasing magnitude in China was particularly pronounced, leading to a more significant decline in dominant strains such as G9P[8] in recent years. In terms of Shenzhen and Zhuhai, their trends exhibited unique regional characteristics (Fig. 1C, F). The G9P[8], which had a significant presence in China, was also the dominant genotype in Shenzhen and Zhuhai between 2020 and 2021. However, in 2022 and 2023, the G8P[8] genotype suddenly became the majority and swept out previous local predominant strains like G9P[8], eventually accounting for a significant proportion of cases, reaching astonishing percentages of 71.43% and 75%, respectively. Although the growth trend of G8P[8] in Shenzhen and Zhuhai may align with that of China, the magnitude of such growth and sweeps in this region was significantly more pronounced.
Phylogenetic analysis of VP7 and VP4
In this study, we conducted phylogenetic analyses of all rotavirus segments for the genetic relationships between our sequences and global RVA strains. Phylogenetic trees on the VP7 and VP4 genes were visualized (Figs. 2 and 3, respectively), with the other trees provided in the supplementary files (Fig. S1).
Fig. 3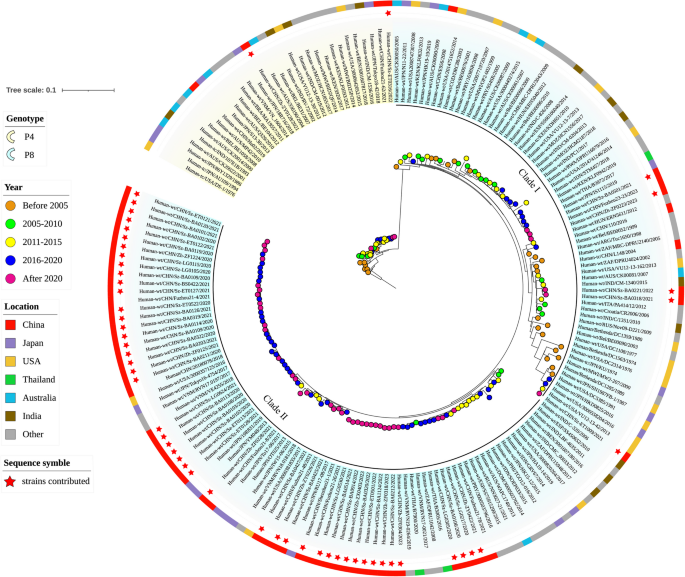
Phylogenetic tree of complete genome sequence of VP4 gene. The background color of the tree represents the genotype, the color strip outside depicts the location, and the color of solid circle represents the time. The strains contributed in this study are indicated by red stars outside. Other sequences can be downloaded from the GenBank database, with accession numbers shown in the Table S2
Fig. 4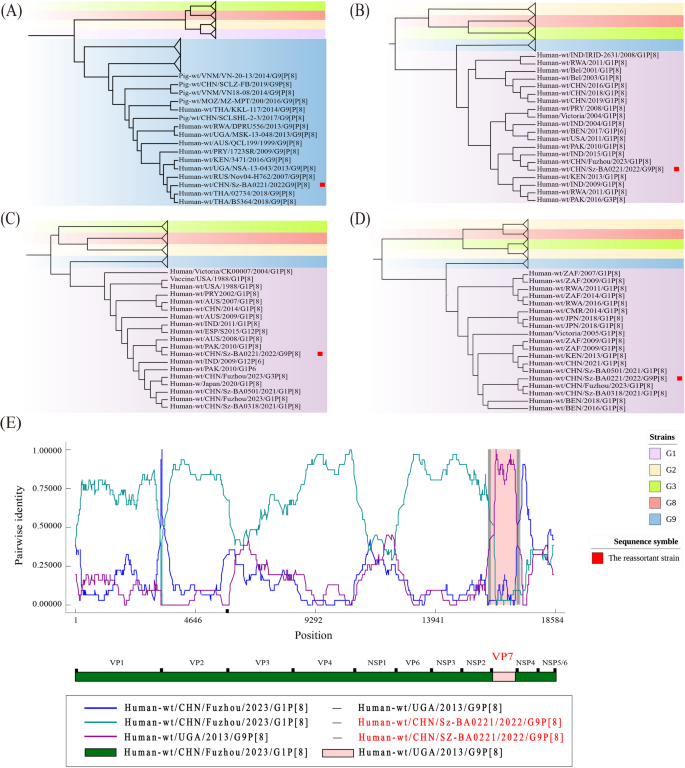
Reassortment analysis of the reassortant strain. Phylogenetic trees were presented for VP7 (A), VP2 (B), VP1 (C), and VP6 (D). The reassortant strain is indicated by red solid square outside. (E). Reassortment results verified by RDP. Reassortant strain name is highlighted in red. The colored lines represent the pairwise similarity between sequences
The phylogenetic tree for VP7 (Fig. 2) contained 53 newly sequenced strains, forming five major genotypes along with other RVA strains: G1, G2, G3, G8, and G9. Among these genotypes, the G9 genotype had the largest number of sequences in Shenzhen and Zhuhai. Hence, the G9 genotype predominated in Shenzhen and Zhuhai in 2020 and 2021, which is consistent with our previous observation (Fig. 1). This G9 genotype can be further divided into three evolutionary clades: Clade I, Clade II and Clade III. Among those clades, the majority of our contributed strains were clustered in Clade II, demonstrating a strong genetic relationship with strains in 2021 from Fuzhou, China. The remaining contributed strains were closely related to some strains reported in Japan in 2018. Within Clade III, most of the contributed strains exhibited a close genetic relationship with strains found in Fuzhou and Hong Kong, China, with one exceptional strain being far from others. Besides the G9 genotype, our contributed sequences were also widely distributed in the G8 genotype. Notably, these strains were clustered together and were genetically related to viral strains from Fuzhou and Wuhan during 2011–2022.
Moreover, we find some interesting phenomenon through the temporal distribution on the phylogenetic tree of VP7 (Fig. 2). Within the G9 genotype, all the strains in Clade I were relatively early, being before 2010. In Clade II, which contains the most newly sequenced strains, strains had a temporal distribution mostly after 2016, indicating that these strains have only been discovered in recent years. Clade III was an intermediate between Clade I and Clade II, evidenced by the presence of both older strains and recent ones. Distinct from the G9 genotype, the majority of G8 strains were generally recent. This phenomenon corresponds with our previous observation, indicating that G8 is an emerging novel genotype.
The phylogenetic tree for VP4 (Fig. 3) contained 54 newly sequenced strains and primarily demonstrated two major genetic lineages, P[4] and P[8]. Generally, P[8] comprised more than half of the tree, consistent with the previous report that P[8] was the predominant circulating rotavirus globally [34]. Within the P[8] branch, two evolutionary clades were formed, namely Clade I and Clade II. Clade II contains a significant number of prototype strains with high internal sequence similarity, clustered together on the phylogenetic tree, and are closely related to strains from Japan and Fuzhou, China. Clade I consists of P[8] strains that show genetic relationships similar to those found in India, USA, and Fuzhou, China. In terms of our contributed strains in P[8] genotype, most of them were distributed in Clade II, with only a few strains being classified into Clade I. In contrast to the P[8] genotype, P[4] only accounted for a small part of the evolutionary tree (Fig. 3). Similarly, the P[4] genotype only contains two strains contributed in this research. within the P[4] lineage are from Shenzhen and Zhuhai respectively. The G2P[4] strain from Shenzhen exhibits a close genetic relationship with strains from Fuzhou, China, Japan, and India, whereas the G2P[4] strain from Zhuhai shares a similar evolutionary relationship with strains from Australia, Japan, and Vietnam.
One intriguing phenomenon is that the lineages within the P[8] genotype can exhibit distinct temporal and regional distribution characteristics. Generally, the P[8] genotype consists of two major Clades: Clade I and Clade II. In Clade I, most of the strains date back to before 2015, with a relatively dispersed regional distribution, predominantly in the United States, India, China, and Australia. Notably, the evolution of this branch might come to an abrupt end without further development, instead giving rise to a new clade. On the contrary, most strains in Clade II were dated after 2016, suggesting that this group have been emerging in the past decade. In terms of the regional distribution, this clade was mainly distributed in East Asia, with the newly sequenced strains from China and closely related strains from Japan. This significant temporal and geographic differences between two P[8] evolutionary clades may indicate their different potential evolutionary directions of these clades.
Reassortment analysis of RVAs
In this study, we identified a genetic reassortment in a sequenced strain. This reassortment segment (PV943222, Fig. 4) was collected from Shenzhen in 2022. According to the phylogenetic trees, the VP7 segment of this strain was classified into the G9 genotype (Figs. 2 and 4A, VP7 segment), whereas the other segments were observed to be more closely related to corresponding segments of the G1 genotype. We selected three representative segments summarized in the reassortment analysis (Fig. 4B to D), with the complete phylogenetic trees of these nine segments presented in Fig. S1.
To further verify this reassortment, we choose the reassortment strain and its genetically related strains for further investigations. For each strain, we concatenated the genomic segments from 1 to 11 into a whole-genome sequence. Then, we conducted RDP analysis on those whole-genome sequences for recombination detections (Fig. 4E). A segment exchange was observed at VP7, in which the breakpoint of this genetic exchange was precisely located within the full-length region of the VP7 gene. Therefore, this result confirmed the occurrence of a reassortment event, in which the major parent was a G1P[8] strain from China, in 2022, and the minor parent was a G9P[8] strain from Uganda in 2013.
Recombination analysis of RVAs
Based on previous research [11,12,13]recombination is considered one significant driver of viral evolution. Consequently, we conducted a recombination analysis on all the 11 rotavirus gene segments by RDP4 (Fig. 5). The sequences used for recombination analysis included the remaining global RVA sequences and our own contributed sequences. We primarily focused on recombination events related to our newly sequences from the Shenzhen and Zhuhai. Overall, recombination events associated with our 57 newly sequenced strains included two cases.
Fig. 5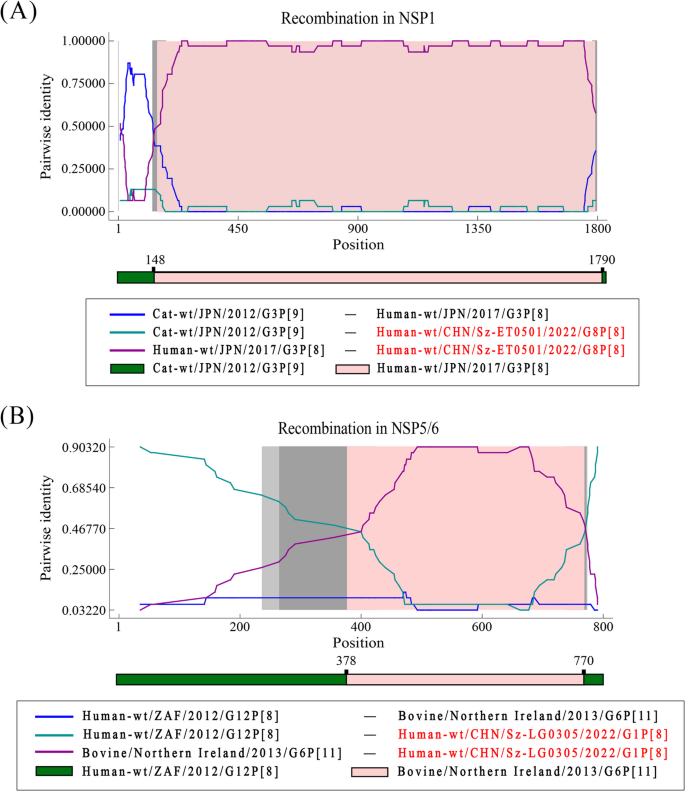
Recombination analysis of rotavirus in NSP1 and NSP5/6. A. Event in NSP1. B Event in NSP5/6. The green line compares the minor parent to the recombinant, purple line compares the major parent to the recombinant, and the blue line compares the major parent to the minor parent. The gray areas represent the degree of confidence. Recombinant strain names are highlighted in red
One recombination event occurred in the NSP1 gene of the G8P[8] strain from Shenzhen (PV948568). The major parent was a G3P[9] strain from Japan in 2012 (LC790348), and the minor parent was a G3P[8] strain from Japan in 2017 (LC542388). The recombination breakpoints were located at 148 bp and 1790 bp, covering almost the entire fifth segment, which encodes the NSP1. The other recombination event was identified in the NSP5/6 gene of the G1P[8] strain (PV943291), also from Shenzhen. the major parent was a G12P[8] strain from South Africa in 2012 (KJ752360), and the minor parent was a G6P[1] strain from Ireland in 2017 (OL988992). The recombination breakpoints were found to be at 378 bp and 770 bp. Interestingly, the two fragments in the recombinant strain are approximately of the same length. Therefore, the genomic contribution of the major parent and the minor parent to the recombinant strain is nearly identical.
We also discovered a noteworthy phenomenon from a geographical perspective. The recombinant strains on NSP1 gene are derived from two Japanese strains, which is relatively reasonable considering Japan’s geographic proximity to China. However, for another recombinant strain on NSP5/6 gene, its recombination fragments are derived from a South African strain and an Irish strain, which can be vastly rare considering their geographic distances.