
Study design and ethical approval
This randomized, blinded, controlled trial was conducted at First People’s Hospital of Yichang (The People’s Hospital of China Three Gorges University), approved by its Ethics Committee on December 12, 2022 (PJ-KY2022-35), and prospectively registered in the Chinese Clinical Trial Registry (ChiCTR2300073162). Patients were recruited from January 2023 to September 2023. Written informed consent was obtained from all participants after a thorough explanation of the study objectives, procedures, potential risks, and benefits. Participation was entirely voluntary. The study was conducted in strict accordance with research ethics guidelines, with particular emphasis on the protection of participant rights. Strict data confidentiality measures were implemented to ensure privacy and data security. All participants retained the right to withdraw from the study at any time without discrimination or negative consequences.
Inclusion and exclusion criteria
The inclusion criteria were as follows: female patients aged 45 to 65 years, scheduled to undergo total laparoscopic hysterectomy, classified as ASA physical status I or II, and with a body weight between 50 and 80 kg.
Exclusion criteria included: uncontrolled or untreated major systemic diseases (such as hypertension, heart disease, or diabetes); history of neck surgery or shoulder pain-related diseases; severe hepatic, renal, or pulmonary dysfunction; long-term use of analgesics, sedatives, or anticoagulant/antiplatelet agents (e.g., warfarin, DOACs, clopidogrel); known coagulopathy (INR > 1.4 or platelet count < 100 × 109/L); and inability to comply with the study protocol.
Grouping and interventions
A total of 198 eligible patients were prospectively enrolled following preoperative evaluation by the anesthesia and surgical teams. The 198 participants were randomly assigned, using a random number table, into six groups (A1, A2, A3, B1, B2, B3), with 33 patients in each group (Fig. 1). Allocation information was sealed in opaque envelopes by an independent investigator and revealed by the circulating nurse after anesthesia induction to inform the attending anesthesiologist.
Fig. 1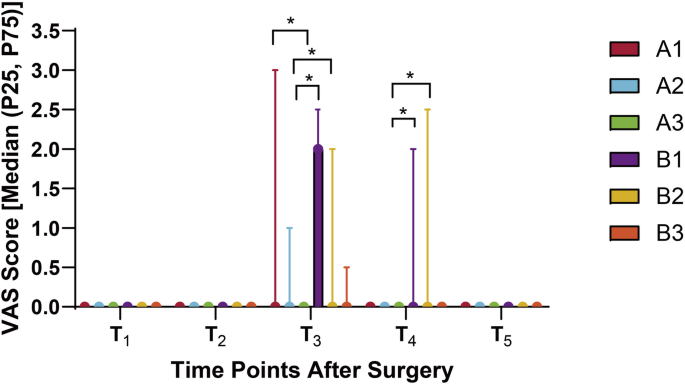
Randomized study flowchart for laparoscopic total hysterectomy
All patients underwent ultrasound-guided right phrenic nerve block. The procedure was performed with the patient in the supine position and the head slightly turned to the left. A high-frequency linear probe was used to identify anatomical structures, including the anterior scalene muscle (ASM) and middle scalene muscle (MSM). After skin disinfection, the phrenic nerve was located on the surface of the anterior scalene muscle at the level of the sixth cervical vertebra. Using an in-plane technique, a 25-G, 6-cm needle was advanced laterally towards the phrenic nerve (adjacent to the brachial plexus). Upon confirming the needle tip position, a syringe with an extension tube was connected, and 6 mL of the study solution was injected slowly (Fig. 2).
Fig. 2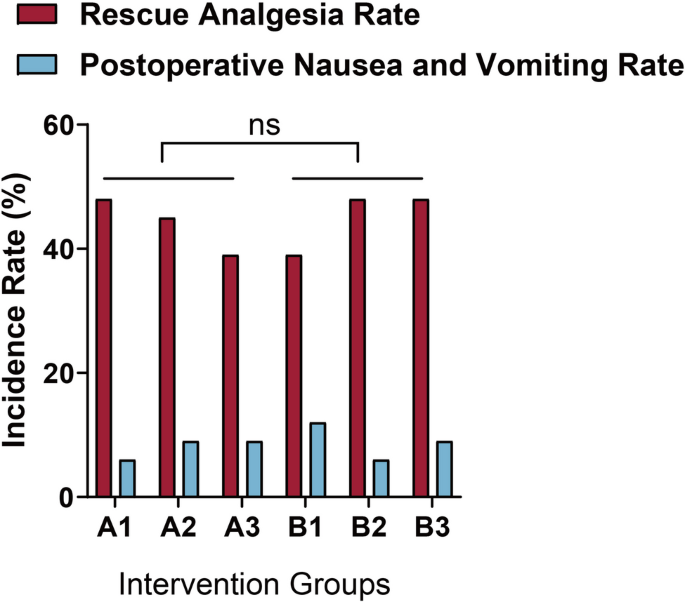
Illustration of injection procedure. Note: ASM, anterior scalene muscle; MSM, medial scalene muscle; → brachial plexus; →▲ phrenic nerve
The timing of the block and the concentration of ropivacaine were as follows: In groups A1, A2, and A3, phrenic nerve block was performed after anesthesia induction and before CO2 pneumoperitoneum, using 0.25%, 0.3%, and 0.375% ropivacaine, respectively. In groups B1, B2, and B3, the block was performed at the end of surgery (during skin closure), with the same respective concentrations of ropivacaine. The same experienced anesthesiologist performed all blocks. For groups A1 to A3, CO2 pneumoperitoneum was established 20 min after the block (i.e., 20 min after the injection, when the block had fully taken effect), with intra-abdominal pressure maintained at 12–13 mmHg. The entire procedure was performed using a no-touch technique. Skin antisepsis was achieved with an ethanol solution containing 2% chlorhexidine gluconate. The ultrasound probe was covered with a sterile disposable sheath, and the operator wore sterile gloves and used sterile coupling gel [26].
Anesthesia protocol
All procedures were performed by the same experienced anesthetist under ultrasound guidance. The patient was positioned supine with a slight leftward head turn. A high-frequency linear array probe was used to scan the anatomical structure and plan the needle path. Following skin antisepsis, the target neural structure was identified under ultrasound guidance. After drug preparation, a 25-gauge, 6-cm needle was inserted in-plane from the lateral side and advanced toward the phrenic nerve. The syringe was connected via an extension line, and 6 mL of ropivacaine, prepared at rigorously controlled concentrations, was slowly administered (Fig. 2). Concentration selection was based on prior evidence indicating that 0.25% represents the lower threshold for diaphragmatic paresis risk [22].
In groups A1, A2, and A3, the artificial CO2 pneumoperitoneum was established 20 min after the full effect of ropivacaine, maintaining an intra-abdominal pressure of 12–13 mmHg. Patients fasted from the day before surgery. Peripheral venous access was established upon entering the operating room, and blood pressure, oxygen saturation, and an electrocardiogram were monitored. Anesthetic induction was achieved with intravenous midazolam (0.075 mg/kg), propofol (2 mg/kg), sufentanil (0.3 µg/kg), and rocuronium (0.6 mg/kg). Mechanical ventilation was initiated post-intubation, with a tidal volume of 8 mL/kg, a ventilation rate of 12 breaths per minute, an I: E ratio of 1:2, and an inspired oxygen concentration of 100%. During surgery, propofol (4–5 mg/kg/h), remifentanil (0.1–0.2 µg/kg/min), and dexamethasone (0.06 mg/kg/h) were administered intravenously. End-tidal CO2 pressure was maintained at 35–45 mmHg, and systolic blood pressure and heart rate fluctuations did not exceed 20% of baseline values. Crystalloid and colloid fluids were administered intravenously during surgery. Dexamethasone administration ceased 30 min before the end of surgery. Flurbiprofen axetil (50 mg) was administered intravenously 10 min before surgery for analgesia, and propofol and remifentanil were discontinued postoperatively. Flumazenil (50 mg), atropine (0.01 mg/kg), and neostigmine (0.02 mg/kg) were administered intravenously to reverse anesthesia. Postoperatively, patients received rescue analgesia with tramadol (30 mg) when the visual analog scale (VAS) score for abdominal incision pain reached 4 or higher.
Postoperative Follow-up and outcome measures
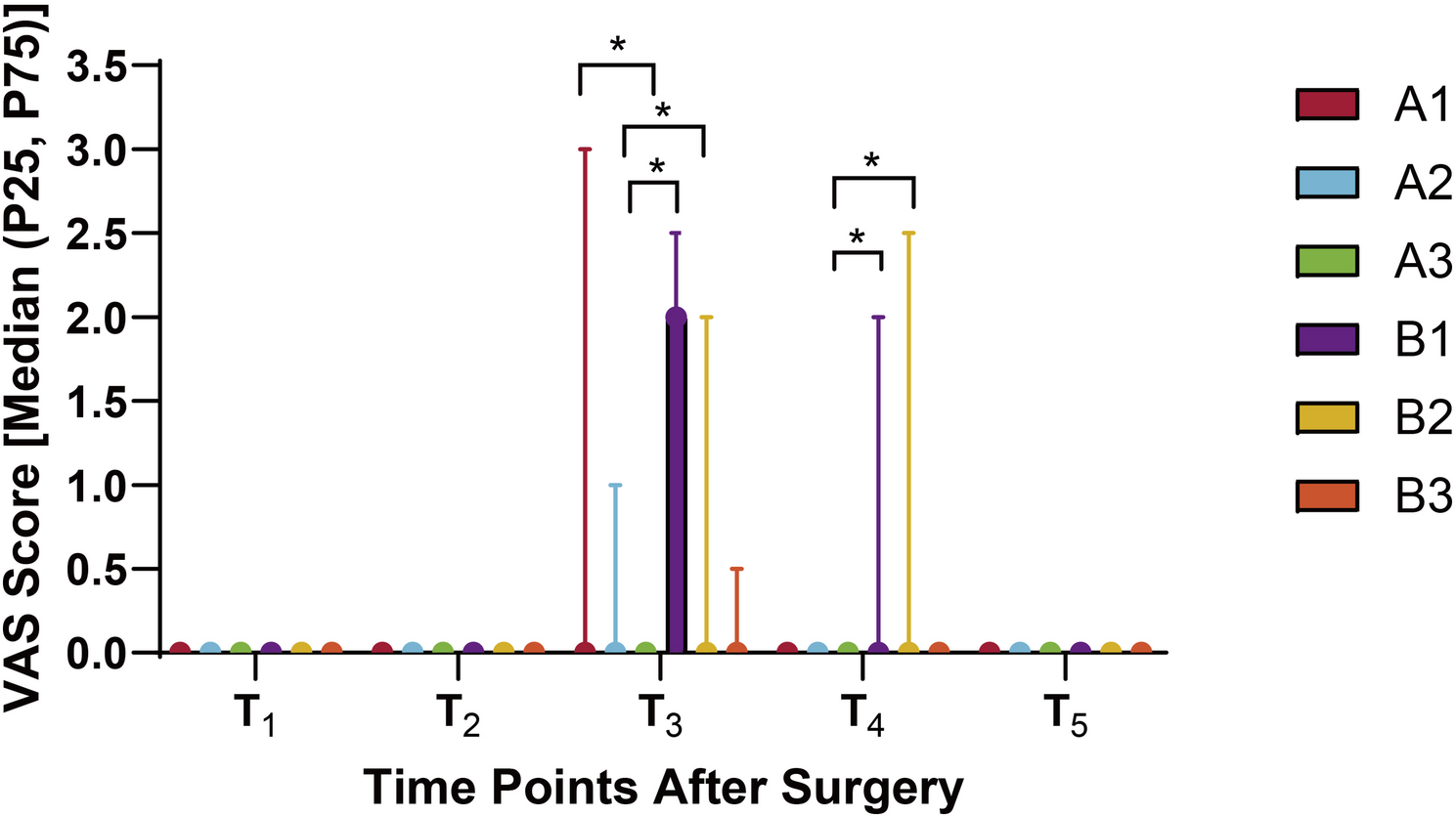
Detailed follow-ups were conducted on the six patient groups at postoperative intervals of 0.5 h (T1), 6 h (T2), 24 h (T3), 48 h (T4), and 72 h (T5). Each participant completed all five repeated measurements. Primary outcomes included the incidence and severity of PLSP at each time point, typically assessed using the VAS or Numeric Rating Scale (NRS). Pain intensity was measured using the VAS. Respiratory rate (RR) and peripheral oxygen saturation (SpO2) were recorded every 30 min during the first 6 h postoperatively, with RR > 20 breaths/min or SpO2 < 94% defined as abnormal thresholds [26]. Baseline characteristics included age, body mass index (BMI), and American Society of Anesthesiologists (ASA) classification. Intraoperative parameters included pneumoperitoneum duration, sufentanil dosage, and propofol dosage. Postoperative variables included extubation time, rescue analgesia rate, incidence of postoperative nausea and vomiting (PONV), and any episodes of persistent dyspnea or oxygen therapy requirement.
Sample size estimation and statistical analysis
Sample size estimation was performed using G*Power 3.1 software. A mixed-model analysis of variance (ANOVA) was planned to evaluate the effects of intervention timing, group allocation, and their interaction. The statistical model was based on an F-test designed for a 6-group × 5-time-point repeated-measures ANOVA, accounting for both between- and within-subject factors. The correlation between repeated measures within each subject was assumed, and sphericity was assessed using Mauchly’s test. When the sphericity assumption was violated (P < 0.05), degrees of freedom were adjusted using the Greenhouse–Geisser correction. All tests were two-tailed with a significance level of α = 0.05.
Due to the absence of pilot data, a conservative medium effect size (f = 0.25) was adopted based on prior studies investigating the effect of regional nerve blocks on VAS scores [23], consistent with Cohen’s definition of a medium effect. The statistical power (1-β) was set at 80%, and α at 0.05. Based on these parameters, the minimum sample size was calculated as 180 patients (30 per group). To compensate for potential data loss (e.g., dropout, protocol deviation), an additional 10% (18 patients) was included, resulting in a final sample size of 198 patients (33 per group). This adjustment aligns with the CONSORT guidelines for sample size planning in randomized trials [27].
Clinical data were analyzed using SPSS version 22.0. Continuous variables were expressed as mean ± standard deviation (SD), and normality was tested using the Shapiro-Wilk test. For normally distributed data, intergroup comparisons were conducted using independent-sample t-tests (two groups) or one-way ANOVA (multiple groups); for non-normal distributions, the Mann–Whitney U test or Kruskal-Wallis test was used. Categorical variables were presented as frequencies (n) and percentages (%), and compared using the chi-square (χ2) test or Fisher’s exact test.
For multiple comparisons, both raw and Bonferroni-adjusted P-values were reported. Bonferroni correction was applied by multiplying the raw P-value by the number of comparisons (k), i.e., adjusted P = raw P × k. A Bonferroni-adjusted P < 0.05 was considered statistically significant. A two-sided P < 0.05 was regarded as indicative of statistical significance throughout all analyses.