
PacBio Revio HiFiPCR-free Revio HiFi Sequencing (Dataset ID: PB-HiFi-1)
Library Preparation
Library preparation was performed using previously isolated genomic DNA (gDNA), as described above in “Tumor and normal samples for measurement”, from non-viable HG008-T cells (batch 0823p23) and non-viable HG008-N-P tissue. Sizing of the isolated gDNA was performed prior to library preparation to confirm the gDNA was of sufficient length for long-read sequencing. The average sizing of smear analysis from Pulsed Field Gel Electrophoresis (PFGE) with an Femto Pulse System (Agilent) was approximately 58 kbp for HG008-T and 29 kbp for HG008-N-P.
Library preparation was performed following PacBio procedure and checklist: “Preparing whole genome and metagenome libraries using SMRTbell® prep kit 3.0” (PN: 102-166-600 REV02 MAR2023). This protocol uses the PacBio SMRTbell prep kit 3.0 (PN: 102-182-700), SMRTbell adapter index plate 96 A (PN: 102-009-200), and SMRTbell cleanup bead kit (PN 103-306-300). Briefly, 6.4 ug of HG008-T and 6.1 ug of HG008-N-P were sheared separately on a Megaruptor 3 (Diagenode, B06010003) to a target size of 15 to 20 kb. Two library preparations were performed, one using sheared gDNA inputs of 3.1 ug of HG008-T and one using 4.1 ug of HG008-N-P. Sheared samples were subjected to a combined DNA repair and A-tailing reaction followed by ligation of sequencing adapters to create SMRTbell libraries. SMRTbell libraries were treated with a nuclease mix to remove unligated library components and then size-selected on a Sage Science PippinHT system using a 0.75% agarose gel cassette with cassette definition file, “6–10 kb High Pass Marker 75E” to remove fragments less than 10 kb.
Whole Genome Sequencing
PacBio SMRTbell libraries were prepared for HiFi sequencing (i.e. circular consensus sequencing)17 by annealing and binding sequencing primers and polymerase, respectively, to the SMRTbell templates using Revio polymerase kit (PN 102-817-600) and following instructions provided in the Sample Setup module of PacBio SMRT Link software (v13.0), a web-based end-to-end sequencing workflow manager. Sequencing-ready SMRTbell libraries were sequenced on a Revio system using the following reagents and consumables: Revio sequencing plate (PN: 102-587-400) and Revio SMRT Cell tray (PN: 102-202-200). 24-hour movies were run. Two SMRT Cells were run for HG008-T and one SMRT Cell for HG008-N-P.
Validation Methods
The PacBio Revio unaligned bams were aligned to the GRCh38 reference genome using the PacBio pbmm2 aligner as part of the PacBio HiFi-human-WGS_WDL (germline) pipeline v1.1.0 (https://github.com/PacificBiosciences/HiFi-human-WGS-WDL). Cramino, Nanoplot and Samtools stats tools were used to assess the quality of the alignments (Table S2, see Supplementary Information document). A summary of QC metrics can be found Table 4. While the method used maintains base modification (methylation) information, this was not assessed as part of the data validation process.
PCR-free Revio HiFi Sequencing (Dataset ID: PB-HiFi-2)
DNA Isolation
Genomic DNA (gDNA) was isolated from approximately five million non-viable HG008-T cells (batch 0823p23) and 40 mg of non-viable HG008-N-D tissue using the Revvity chemagic Prime 8 Instrument. Cells were resuspended and homogenized in the tissue lysis buffer, vortexed, and treated with RNase. The homogenate was then transferred to the chemagic Prime 8 instrument where DNA was extracted using the chemagic Prime DNA Blood 2k Kit H24 kit (PN: CMG-1497). For the normal tissue sample, the tissue was minced with a razor blade, incubated overnight in the lysis buffer with proteinase K. The following day the homogenate was subjected to an RNase treatment and transferred to the chemagic Prime 8 instrument where DNA was extracted using the chemagic Prime DNA blood extraction kit (CMG-1497). Genomic DNA mass yields for tumor and normal samples were approximately 68 and 72 ug, respectively. Resulting DNA size was measured on an Agilent Femto Pulse System with average sizing of approximately 81 kb for gDNA from tumor cell line and 59 kb for gDNA from normal duodenal tissue.
Library Preparation
Genomic DNA (7.5 ug) was sheared using Covaris g-tubes to achieve an average size of 15 to 20 kb. Sheared DNA was size selected with a Sage Science PippinHT using the 15 to 20 kb high-pass 75E cassette definition (gel cassette HPE7510) to eliminate fragments smaller than 15 kb. One library for each sample type was prepared. The PacBio procedure for library preparation was performed using the PacBio SMRTbell prep kit 3.0 (PN: 102-182-700) and the SMRTbell adapter index plate 96 A (PN: 102-009-200) for repair and A-tailing, adapter ligation and nuclease treatment to remove unligated fragments. Final libraries were purified using 1X PacBio SMRTBell Cleanup beads, eluted in 30 µL PacBio elution buffer and quantified using the Qubit dsDNA quantification high-sensitivity assay (Thermo Fisher Scientific). Final library size was determined using the Agilent Femtopulse.
Sequencing Methods
PacBio SMRTbell libraries were prepared for HiFi sequencing (i.e. circular consensus sequencing) by annealing and binding sequencing primers and polymerase, respectively, to the SMRTbell templates using Revio polymerase kit (PN: 102-817-600) and following instructions provided in the Sample Setup module of PacBio SMRT Link software (13.0.0.207600). Sequencing-ready SMRTbell libraries were sequenced on a PacBio Revio system (ICS version 13.0.0.212033). 30-hour movies were run. Two SMRT cells each were run for HG008-T and HG008-N-D. QC metrics from SMRTlink software including HiFi reads, mean read length, P1%, etc were as expected.
Validation Methods
The PacBio Revio sequencing unaligned bams raw data were aligned to the GRCh38 reference genome using the PacBio pbmm2 aligner as part of the PacBio Somatic variant calling Pipeline v0.7 (https://github.com/PacificBiosciences/HiFi-Somatic-WDL). Cramino, Nanoplot and Samtools stats tools were used to assess the quality of the alignments (Table S2, see Supplementary Information document). A summary of QC metrics can be found Table 4. While the method used maintains base modification (methylation) information, this was not assessed as part of the data validation process.
Oxford Nanopore Technologies PromethIONPCR-free Standard Length PromethION Sequencing (Dataset ID: ONT-std-1)
Library Preparation
Library preparation was performed using previously isolated genomic DNA (gDNA), as described above in “Tumor and normal samples for measurement”, from non-viable HG008-N-P and HG008-N-D tissues. Library preparation was performed with Oxford Nanopore Technologies Ligation Sequencing Kit V14 (PN: SQK-LSK114) using one microgram of gDNA from each tissue.
Whole Genome Sequencing
Nanopore based sequencing was performed on Oxford Nanopore Technologies (ONT) PromethION utilizing ONTs PromethION R10.4.1 Flow Cells (PN: FLO-PRO114M)18. To maximize throughput multiple flow cells were used. Sequencing was performed using four flow cells for the duodenal sample and three flow cells for the pancreatic sample.
Basecalling
The ONT Dorado v0.5.3 basecaller (https://github.com/nanoporetech/dorado) and DNA model, ‘dna_r10.4.1_e8.2_400bps_sup@v4.3.0_5mC_5hmC@v1’.
were used for basecalling, including modified bases. Basecalling successfully completed with no errors. Output from the basecaller is a bam file of unaligned reads including modified bases.
Validation Methods
The Dorado basecaller generates a ‘summary.txt’ file, which was analyzed using PycoQC to assess sequencing quality metrics. The ONT unaligned bams were processed to extract the FASTQ sequences by using samtools then aligned to the GRCh38 reference genome using the Minimap2. Cramino, Nanoplot and Samtools stats tools were used to assess the quality of the alignments (Table S2, see Supplementary Information document). A summary of QC metrics can be found Table 4. While the method used maintains base modification information, this was not assessed as part of the data validation process.
PCR-free Standard Length PromethION Sequencing (Dataset ID: ONT-std-2)
DNA Isolation
DNA was extracted from approximately five million non-viable HG008-T cells (batch 0823p23) utilizing the NEB Monarch® HMW DNA Extraction Kit for Tissue (PN: T3060). Total DNA extracted was approximately 80 µg. Only 5 µg of DNA was sheared using Diagenode’s Megaruptor3 (PN: B06010003) with the DNAFluid + Kit (PN: E07020001) followed by DNA size selection using PacBio SRE kit (PN: 102-208-300). The size of sheared DNA fragments were analyzed on the Agilent Femto Pulse System using genomic DNA 165 kb kit (PN: FP-1002-0275). Fragment size distribution of post-sheared DNA had a peak at approximately 45 kb length.
Library Preparation
The UCSC protocol has been optimized to generate N50s of approximately 30 kb with very high throughput for ligation-based sequencing. Library preparation was performed with Oxford Nanopore Technologies (ONT) Ligation Sequencing Kit XL V14 (PN: SQK-LSK114-XL). Enough library was created for 4 library loads onto flow cells.
Whole Genome Sequencing
Nanopore based sequencing was performed on the ONT PromethION 48/PromethION A-Series Data Acquisition Unit (PN: PRO-SEQ048/PRO-PRCAMP) utilizing ONT PromethION R10.4.1 Flow Cells (PN: FLO-PRO114M). Sequencing was performed on the same day as ligation library preparation. Sequencing was performed utilizing one PromethION R10.4.1 Flow Cell. The flow cell was washed with ONTs Flow Cell Wash Kit XL (PN: EXP-WSH004-XL) each day and re-primed and loaded with fresh library for 4 days of sequencing to maximize throughput.
Basecalling
The ONT Dorado v0.3.4 basecaller (https://github.com/nanoporetech/dorado) and model ‘dna_r10.4.1_e8.2_400bps_sup@v4.2.0/dna_r10.4.1_e8.2_400bps_sup@v4.2.0_5mCG_5hmCG@v2’ were used for basecalling, including modified bases. Basecalling successfully completed with no errors. Output from the basecaller is a bam file of unaligned reads including modified bases.
Validation Methods
The Dorado basecaller generates a ‘summary.txt’ file, which was analyzed using PycoQC to assess sequencing quality metrics. The Oxford Nanopore Technologies sequencing unaligned bams were processed to extract the FASTQ sequences then aligned to the GRCh38 reference genome using the Minimap 2. Cramino, Nanoplot and Samtools stats tools were used to assess the quality of the alignments (Table S2, see Supplementary Information document). A summary of QC metrics can be found Table 4. While the method used maintains base modification (methylation) information, this was not assessed as part of the data validation process.
PCR-free Ultra Long PromethION Sequencing (Dataset ID: ONT-UL-1)
DNA Isolation
Ultra-high molecular weight DNA was extracted from approximately five million non-viable HG008-T cells (batch 0823p23) utilizing New England Biolabs Monarch® HMW DNA Extraction Kit for Tissues (NEB #T3060). Wide bore pipette tips were used throughout to preserve ultralong DNA. No QC of resulting gDNA was performed as viscosity of isolated DNA solution is challenging to work with. Rather ultra long DNA is assessed as part of the Oxford Nanopore Technologies (ONT) sequencing and previous experience with a five million cell as input.
Library Preparation
Library preparation was performed utilizing the ONT Ultra-Long DNA Sequencing Kit Version 14 (SQK-ULK114). The library was eluted in approximately 810 µL volume in order to sequence across additional flow cells to maximize data collection.
Whole Genome Sequencing
Nanopore based sequencing was performed on an ONT PromethION 48/PromethION A-Series Data Acquisition Unit (PN: PRO-SEQ048/PRO-PRCAMP) utilizing ONT PromethION R10.4.1 Flow Cells (PN: FLO-PRO114M). Sequencing was performed the day following library prep utilizing three PromethION R10.4.1 Flow Cells. The flow cells were washed with ONTs Flow Cell Wash Kit XL (EXP-WSH004-XL) each day and reloaded with the library for 4 days of sequencing to maximize throughput.
Basecalling
The ONT Dorado v0.4.3 basecaller (https://github.com/nanoporetech/dorado) and model, ‘dna_r10.4.1_e8.2_400bps_sup@v4.2.0/dna_r10.4.1_e8.2_400bps_sup@v4.2.0_5mCG_5hmCG@v2’ were used for basecalling, including modified bases. Basecalling successfully completed with no errors. Output from the basecaller is a bam file of unaligned reads including modified bases.
Validation Methods
The Dorado basecaller generates a ‘summary.txt’ file, which was analyzed using PycoQC to assess sequencing quality metrics.The ONT unaligned bams were processed to extract the FASTQ sequences by using samtools then aligned to the GRCh38 reference genome using the Minimap2. Cramino, Nanoplot and Samtools stats tools were used to assess the quality of the alignments (Table S2, see Supplementary Information document). A summary of QC metrics can be found Table 4. While the method used maintains base modification (methylation) information, this was not assessed as part of the data validation process.
PCR-free Ultra Long PromethION Sequencing (Dataset ID: ONT-UL-2)
DNA Isolation
Ultra-high molecular weight DNA was extracted from approximately five million non-viable HG008-T cells (batch 0823p23) utilizing New England Biolabs Monarch® HMW DNA Extraction Kit for Cells (NEB #T3050). Extraction yielded ~25-30 ug with DNA hundreds of kilobases long, with some exceeding megabases, as measured by preliminary analysis of Oxford Nanopore Technologies (ONT) sequencing.
Library Preparation
A single library preparation was performed utilizing the ONT Ultra-Long DNA Sequencing Kit Version 14 (SQK-ULK114) using all isolated gDNA.
Whole Genome Sequencing
Nanopore based sequencing was performed on an ONT 48/PromethION A-Series Data Acquisition Unit (PN: PRO-SEQ048/PRO-PRCAMP) utilizing ONTs ULK114 chemistry with the E8.2.1 motor protein and PromethION R10.4.1 Flow Cells (PN: FLO-PRO114M). Two flow cells were used and received a total of four library loads with three wash (ONT Flow Cell Wash Kit XL PN: EXP-WSH004-XL) and reload steps per flowcell.
Basecalling
The ONT Dorado v0.8.1 basecaller (https://github.com/nanoporetech/dorado) and the super accuracy (sup) model (v5.0.0), were used for basecalling, including 5mC_5hmC modification calls (v2.0.1). Basecalling successfully completed with no errors. Output from the basecaller is a bam file of unaligned reads including modified bases.
Validation Methods
The ONT unaligned bams were processed to extract the FASTQ sequences by using samtools then aligned to the GRCh38 reference genome using the Minimap2. Cramino, Nanoplot and Samtools stats tools were used to assess the quality of the alignments (Table S2, see Supplementary Information document). A summary of QC metrics can be found Table 4. While the method used maintains base modification information, this was not assessed as part of the data validation process.
Single cell whole genome sequencingBioSkryb ResolveDNA with Illumina Low Pass Sequencing (Dataset ID: sc-ILMN-1)
Whole Genome Amplification (WGA)
Approximately two million non-viable HG008-T cells (batch 0823p23) were sorted with a Sony SH800 using a 130 micron chip. Singlet (FSC-A/FSC-H, BSC-A/BSC-W) and live-cell (PI negative, top 70% Calcein-AM positive) gating was employed for single cell sorting into a 384-well plate with 3 µL of BioSkryb Cell Buffer (PN: 100199). Individual cells were processed within wells using the ResolveDNA WGA Kit (PN: 100136) following steps outlined in the ResolveDNA Whole Genome Amplification Kit v2.0 User Guide. Primary Template Directed Amplification (PTA) to amplify single cell whole genomes and transform into Illumina-sequenceable libraries19. Approximately 70% of cells met specifications for BioSkryb WGA metrics. Dual-indexed Illumina-compatible libraries were generated for the top performing 120 cells.
Illumina Low-Pass Sequencing
Illumina low pass sequencing performed to QC the single cell libraries. libraries were pooled and sequenced on a single flowcell targeting 2 million reads as an initial QC using an Illumina NextSeq. 1000 instrument with 2 × 50 paired-end reads with P2 reagents v3 (PN: 200446811).
Single Cell Library Validation
Single-cell libraries were assessed by running BioSkryb’s BJ-DNA-QC pipeline (https://docs.basejumper.bioskryb.com/pipelines/secondary/bj-dna-qc/docs/) through the BioSkryb analytics platform, BaseJumper. The BJ-DNA-QC pipeline uses the Illumina low-pass sequencing data and generates several QC metrics that help assess whether the single-cell libraries are ready for high-depth sequencing (Table S2, see Supplementary Information document). Briefly, reads were aligned to the GRCh38 reference and QC-metrics obtained from the derived alignment and are used to censor single-cells. QC metrics, used for validation, are further discussed in the Technical Validation section.
BioSkryb ResolveDNA with Ultima High Throughput Sequencing (Dataset ID: sc-Ultima-1)
Library Conversion
BioSkryb Genomics previously prepared 120 ResolveDNA single cell libraries for Illumina low-coverage sequencing; described above for the sc-ILMN-1 dataset. For high-coverage Ultima sequencing these libraries were then converted to a format compatible with the Ultima Genomics sequencing platform (UG 100), following the Ultima Genomics Library Conversion protocol in UG Library Amplification Kit v3 User Guide (Publication #: P00025 Rev. B). Library Amplification followed option 2 for starting with non-UG adapters. The UG Library Amplification Kit v3 (supplied by Ultima Genomics) and the IDT kit xGen™ Indexing Primers for Ultima (PNs: 10016992 and 10016993) were used for conversion and enabled multiplexing of the single cell libraries. The library amplification step was modified to replace the UG Index Primer and UG Universal primer with 5.0 µL of primer mix from the appropriate well of the IDT index primer plate. Ten nanograms of input library was used for each reaction and seven cycles of PCR were performed per the amplification protocol. Final converted libraries were QC’d for size and yield using an Agilent Tapestation and Qubit, respectively. Two plates of 120 individual Ultima single-cell libraries were sent to Ultima Genomics for sequencing.
Whole Genome Sequencing
Sequencing of the converted (Illumina to Ultima) libraries was performed on the UG 100 (software version: APL 5.1.0.19) using V27 Chemistry. Single end, 300 bp reads were generated using the UG_116cycles_Baseline_1.5.3.2 sequencing recipe and flow order [T,G,C,A] on a single wafer. A human genome control sample HG002 (Coriell Nucleic acid ID: NA24385) was spiked into each run in order to ensure quality of the samples being sequenced.
Validation
To generate sequencing measures and QC statistics, a custom Ultima tool was run that implements select picard metrics (https://github.com/broadinstitute/picard). This tool collects the metrics on board of the UG100 sequencing platform in parallel with producing sorted, duplicate-marked GRCh38 aligned CRAM files. Alignments were produced using an Ultima-optimized version of BWA-MEM (Table S2, see Supplementary Information document). QC metrics, used for validation, are further discussed in the Technical Validation section.
Whole genome optical mappingOptical Mapping with Bionano Saphyr (Dataset ID: Bionano-1)
DNA Isolation
Approximately two million non-viable HG008-T cells (batch 0823p23), were washed with Bionano Stabilizing Buffer (PN: 20394 and 20397) by MGH at the time of pelleting and freezing. Upon receipt of the cell pellet, ultra-high molecular weight genomic DNA (gDNA) was isolated using the Bionano SP-G2 Blood & Cell Culture DNA Isolation Kit (PN: 80060) with a yield of approximately 6.5 ug of gDNA (100 ng/µL in 65 µL elution) with an N50 greater than 150 kbp.
Sample Preparation
Sequence-specific labeling of the gDNA was performed using the Bionano Direct Label and Stain-G2 (DLS-G2) Kit (PN: 80046).
Optical Genome Mapping
Labeled DNA was loaded onto a Bionano Saphyr Chip G3.3 and measured with the Saphyr System (ICS v5.3.23013.1). Molecules and labels imaged on the Saphyr instrument were digitized and saved in a BNX format (raw data) (https://bionanogenomics.com/wp-content/uploads/2018/04/30038-BNX-File-Format-Specification-Sheet.pdf) which served as input into downstream analyses. The Rare Variant Pipeline (RVP) (https://bionano.com/wp-content/uploads/2024/04/CG-30375-Analysis-Quick-Start-Annotated-Rare-Variant-Structural-Variant-Calling.pdf) detected SVs by the “split-read” analysis, where initial molecule alignment and molecule extension refinement allows identification and detection of somatic variants, and identifies variants that are 5 kbp or longer in size. The Access software (v1.8) was used to generate high confidence callsets for SVs and unique SVs not present in the Bionano internal control database. High confidence CNV calls were generated after recentering based on the hypodiploid tumor genome. Regions where unique SV and CNV calls overlap with Bionano custom pan cancer gene list are also reported.
Validation
This sample was processed based on manufacturer protocol20 (Table S2, see Supplementary Information document). QC metrics, used for validation, are further discussed in the Technical Validation section.
Cytogenetic analysisG-Banded Karyotyping (MGH p31 karyotyping-1 and NIST p18 karyotyping-2)
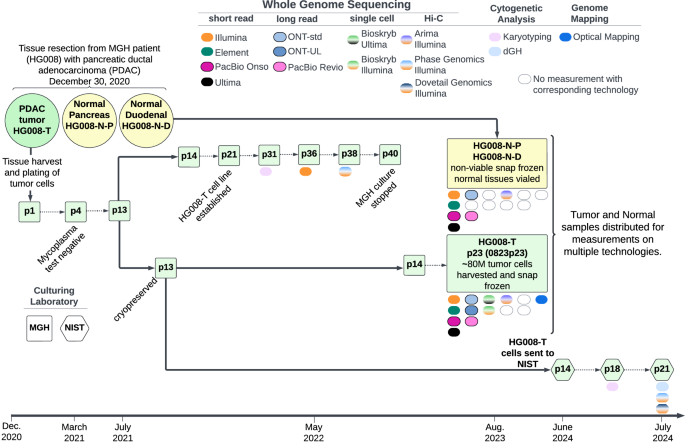
Karyotyping was performed by KaryoLogic Inc (Durham, NC) for two passages of HG008-T as shown in Fig. 2. Approximately one million viable HG008-T cells were plated in T25 cell culture flasks, two from MGH and one from NIST, and sent for karyotyping. Cytogenetic analysis was performed on twenty-five G-banded metaphase spreads of the HG008-T cell line from MGH passage 31. A second analysis was performed in 2024 using viable HG008-T cells from NIST passage 18. Cytogenetic analysis was performed on twenty G-banded metaphase spreads for the HG008-T NIST passage 18 cells (2024).
KROMATID Whole Genome Directional Genomic Hybridization (dGH) (Dataset ID: dGH-1)
Sample Preparation
Sample preparation for dGH analysis was performed at NIST from passage 21 of HG008-T. One million cells were plated in a T25 flask and were cultured overnight. The cell culture medium was replaced with dGH cell prep kit (KROMATID, Cat# dGH-0001) contained culture medium, and additional dose of dGH cell prep kit was added 4 hr after medium replacement. Cells were arrested in the first mitosis with a 4-hour Colcemid (KROMATID, Cat# COL-001) block, harvested, fixed in freshly made fixative [3:1 methanol:acetic acid (Fisher Scientific, Waltham, MA, USA)], and shipped to KROMATID. Metaphase spread preparation along with subsequent UV and exonucleolytic treatments to selectively remove the analog-incorporated daughter strand were performed at KROMATID according to published dGH protocols21.
Directional Genomic Hybridization
For whole genome analysis, KROMATID’s KROMASURE Screen assay was hybridized to the prepared metaphase spreads, then counterstained with DAPI (Vectashield, Vector Laboratories, Newark, CA, USA) and imaged on an Applied Spectral Imaging Harmony system (Applied Spectral Imaging, Carlsbad, CA, performed USA) using a 100X objective. The KROMASURE Screen assay consists of single-stranded, unidirectional, tiled oligos designed to hybridize directly to the unique sequences in each of the 24 human chromosomes. Each chromosome specific complement of oligos is end-labeled with one of five unique spectrally differentiable fluorophores, and each of these “high-density chromosome paints” are combined such that chromosomes painted in the same color can be differentiated by size, shape, and centromere position. Prior to analysis, images of KROMASURE Screen painted metaphase spreads are qualified, processed and sorted into karyograms for rapid, consistent reading of the assay.
A combination of expected KROMASURE Screen signal patterns in the (diploid) human reference genome22, the HG008-T standard G-banding karyotypes, and the copy number variant was used to construct preliminary KROMASURE Screen karyograms in 10 cells, including two (2) genome-doubled cells. The prototype karyograms will be used to generate per-chromosome attribution of inter- and intra-chromosomal structural variation events including inversions, translocations, aneuploidy (gain and loss), insertions, centromere abnormalities and complex events across the sample in additional cells from passage 21 (Fig. 2).
Somatic variant annotation
An initial annotation of a publicly available HG008-T somatic variants called by DRAGEN v.4.2.4 was performed using ANNOVAR (v2020-06-08, https://annovar.openbioinformatics.org/en/latest/)23,24,25. DRAGEN calls were made from the Illumina-PCR-free-1 dataset. ANNOVAR was run using databases and annotation datasets: refGene, ClinVar (v20221231 and v20220320), Catalogue Of Somatic Mutations In Cancer (COSMIC) (v92, https://cancer.sanger.ac.uk/cosmic), gnomAD (v2.1), 1000 Genomes Project (2015 Aug),and the Exome Sequencing Project (ESP), as well as curation of genes commonly mutated in PDAC tumors.
HG008 ancestry analysis
We examined global genetic ancestry of the HG008 tumor and normal pancreatic samples, HG008-T and HG008-N-P (tissue) respectively, utilizing SNVs called by xAtlas from the Illumina-PCR-free-1 dataset. Ancestry was assessed by germline genetic similarity of these “target samples” to reference samples of known genographic origin from the 1000 Genomes Project (Phase 3)26. For computational efficiency, we limited our analysis to chromosome 21. We used PLINK27 to restrict our analysis to SNPs with minor allele frequency greater than or equal to 5% in the reference panel. We then extracted these overlapping loci from the target sample Variant Call File (VCF) and then merged the datasets into a single VCF with bcftools28. During the merging step, we assumed that entries absent from the target sample VCF at these sites were homozygous for the reference allele. We then performed principal component analysis (PCA) using PLINK and visualized genetic similarity on the first three principal component axes.
Haplotype-specific copy number analysis
Haplotype-specific copy number analysis was performed using Wakhan (https://github.com/KolmogorovLab/Wakhan)29. Wakhan computes haplotype-specific coverage of the genome using phased heterozygous germline variants. Then it identifies boundaries of the copy number alteration (CNA) events by analysing SV breakpoints. Prior to running Wakhan, we phased the germline variants with PacBio long-read (Dataset ID: PB-HiFi-1) and Hi-C (Dataset ID: HiC-ILMN-2) data using HiPhase30 and HapCUT231.
STR genotyping
STR genotyping was performed for DNA isolated from the normal pancreatic and duodenal tissues and the HG008-T cells (batch 0823p23 and NIST passage 21) was performed using genomic DNA following a published protocol32. Briefly, STR genotyping via capillary electrophoresis was conducted using PowerPlex Fusion 6 C (Promega) and GlobalFiler (Thermo Fisher Scientific) according to the manufacturer’s protocols. The process targeted 1.0 ng of input DNA and utilized a 3500xL Genetic Analyzer with a 36 cm capillary array and POP-4 polymer (Thermo Fisher Scientific, Cat# A26070). Sample injection was performed at 1.2 kV for 15 seconds. GeneMapper IDX v1.6 (Thermo Fisher Scientific) was employed for data interpretation, using the manufacturer’s provided bins and panels. Alleles were called using an analytical threshold set at 50 RFU. STR typing is provided in Table S4, see Supplementary Information document.