
Mitochondrial diseases are rare genetic disorders that impair mitochondrial oxidative phosphorylation [2]. However, they are some of the most common inherited neurological disorders—in adults, the prevalence of disease, stemming from mutations in both nDNA and mtDNA, is estimated to be 1 in 4300, while the birth prevalence of diseases caused by mtDNA mutation is 1 in 5000 [3]. As with other mitochondrial disorders, patients suffering from primary mitochondrial myopathies (PMM) exhibit diverse clinical phenotypes, including differing symptoms, variable age of onset and severity of disease, stemming from a large number of causative mutations. These features, as well as the lack of a definitive diagnostic standard, pose serious difficulties to clinicians, in establishing the correct early diagnosis [4].
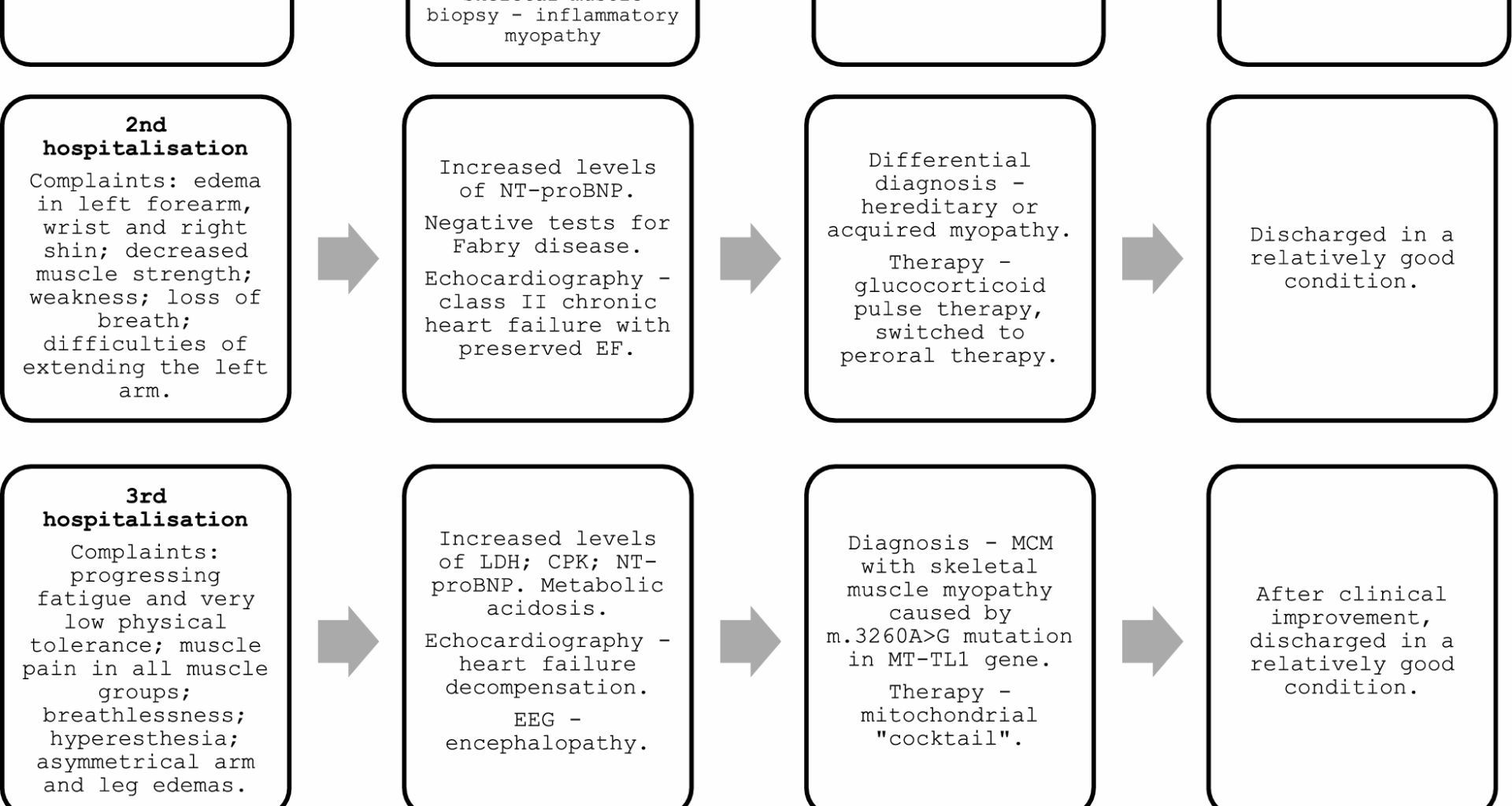
In the case presented in this paper, the process of establishing diagnosis of MCM with skeletal muscle myopathy, caused by a m.3260A > G mutation in MT-TL gene, was challenging. Owing to the rareness of the causative mutation, and, as such—unusual presentation of the symptoms, several diagnoses, including more common ones, such as idiopathic inflammatory myopathy, and more unusual ones, such as Fabry disease, were considered, before arriving at the correct diagnosis. This had led to the subsequent worsening of the patient’s state, owing to the lack of adequate treatment and lifestyle changes.
The MT-TL1 gene (also known as the tRNALeu(UUR) gene) itself is responsible for encoding mitochondrial transfer RNA for leucin (tRNALeu) which adds leucine to the polypeptide chain of mtDNA-encoded subunits during translation [9].
As previously mentioned, mitochondrial myopathies can present with a diverse set of clinical features. Highly varied phenotypical presentations frequently lead to diagnostic dilemmas and are among the most serious challenges in the process of diagnosis and treatment of any mitochondrial disorder [2, 10]. Clinical features can include a wide range of manifestations, such as various ocular conditions [11], endocrinopathies [12], neurological disorders [13], gastrointestinal [14] symptoms, and others [10]. The skeletal muscle involvement itself is one of the most common features of mitochondrial diseases in general, with symptoms ranging from muscle weakness, exercise intolerance, and exercise induced myalgia, to muscle cramps and muscle wasting [2, 10, 15]. The myopathy usually is slowly progressive and proximal muscles are affected more often; however, it can spread to distal muscles of extremities, as well as muscles of the neck and face [2, 10]. Notably, the presentation of myopathy along with multisystem involvement without a clear origin can be a sign that points to a mitochondrial cause, along with unusual disease severity and observed maternal inheritance pattern [10].
As with the patient presented in this paper, frequent initial complaints include exercise intolerance and progressive muscle weakness, as well as fatigue and muscle pain [16, 17]. Fatigue in particular has been observed as the most common patient-reported symptom [18]. Notably, various frequently seen manifestations that point to a mitochondrial origin of the disease, such as diabetes, hearing loss, as well as ophthalmoplegia and other ocular symptoms [17, 19], were not observed in the patient discussed in this paper. Another frequent manifestation is epilepsy [20], which was observed only a few years after the diagnosis of mitochondrial disease was established.
Patients with mitochondrial myopathy can suffer from a recognized clinical syndrome, such as chronic progressive external ophthalmoplegia (CPEO), Leigh syndrome, mitochondrial encephalopathy and lactic acidosis with stroke-like episodes (MELAS), neuropathy, ataxia, retinitis pigmentosa (NARP), for example; however, often their illness does not fit into any single specific syndrome, and they present with an oligosymptomatic, or overlapping disease phenotype [19, 21]. According to a study performed by analyzing the North American Mitochondrial Disease Consortium (NAMDC) registry, out of 666 participants, the most frequent syndromes were Leigh syndrome (97 individuals), followed by MELAS (71 individuals), CPEO (55 individuals), and Leber hereditary optic neuropathy (LHON) (28 individuals). However, the most common diagnosis was multisystemic disorder (113 individuals), not fitting into any classical syndrome [22]. Adult-onset mitochondrial disease generally presents more subtly than pediatric disease. In addition, adulthood onset disease usually does not manifest as a typical clinical syndrome. These cases can either manifest in adulthood for the first time, or, similarly to the case described in this paper, be only recognized in adulthood after a history of nonspecific symptoms starting in childhood [10].
A particularly noteworthy mitochondrial disorder is LHON—a major cause of mitochondrial blindness, affecting approximately 1 in 30 000 individuals, particularly younger patients between the ages of 15 and 35 years. It usually presents with painless bilateral progressive vision loss that rapidly reaches the lowest point in 4–6 weeks. Long-term prognosis is poor—most patients face severe and permanent vision loss (visual acuity worse than 20/400) [11]. It is important to remark that our patient did not have any notable vision impairment that could be attributed to mitochondrial disease.
Usual initial biochemical screening tests include CPK, LDH, lactate, blood glucose, hemoglobin A1c, thyroid, and liver function tests [23]. Elevated plasma lactate is frequently seen in various mitochondrial diseases, including mitochondrial myopathy; however, it can often be only slightly increased, or even normal, especially in adults [19]. However, standard biomarkers are not sufficient for establishing diagnosis, nor disease progression monitoring in adults [24].
If the diagnosis of mitochondrial myopathy is unclear, EMG can be useful to rule out conditions with similar clinical and laboratory findings; however, similarly to the case presented in this paper, often the findings show a nonspecific myopathic pattern, and frequently can be normal [4].
Skeletal muscle biopsy is one of the most important diagnostic tools, and can show histochemical alterations in the skeletal muscle, which indicates mitochondrial dysfunction; however, it can often be unremarkable. Characteristic findings include “ragged red fibers,” “ragged blue fibers,” and COX (cytochrome c oxidase)—negative fibers. Various possible nonspecific findings include internal nuclei, abnormal alterations in fiber size, neural atrophy, and accumulations of glycogen and/or lipids [16]. The timing of the biopsy may depend on various factors. If there is an acute destruction of muscle cells reflected by very high CPK levels, the mitochondrial deficiency findings can be obscured by the overall muscle fiber destruction, thus it may be more informative after the acute episode has subsided [25]. If a genetic disorder would have been suspected initially, our tactics also would prefer first obtaining the genetic results and to seek the morphological diagnosis only if the less invasive testing has come back negative.
Initial cardiovascular testing includes ECG and echocardiography [17]. Hypertrophic cardiomyopathy, dilated cardiomyopathy, and LV noncompaction are the three main manifestations of MCM [26]. It can affect not only myocardium, but also other structures of the heart, such as aortic valves, coronary arteries, cardiac conduction system, or pericardium [27], including the patient discussed in this paper. A common initial finding is progressive diastolic dysfunction and heart failure with preserved EF [7]. It often progresses to LV hypertrophy and systolic dysfunction. Even in the absence of LV hypertrophy, several ECG findings can support the diagnosis of mitochondrial disease, such as short PR interval, T wave abnormalities, and various degrees of atrioventricular block [17, 27]. In fact, in mitochondrial diseases with cardiovascular involvement, minor ECG abnormalities are the most common manifestations [17]. Cardiomyopathy is more common in mitochondrial diseases that present with skeletal muscle myopathy [28]. It is also less commonly seen in adult-onset mitochondrial diseases, whereas 20–40% of children with various mitochondrial diseases develop cardiomyopathy [26, 29]. In total, mtDNA-related cardiomyopathy is estimated to affect at least 1 in 10 000–15 000 of the general population [21].
A fundamental part of diagnosing mitochondrial diseases is genetic counseling, including molecular genetic testing, which can reveal pathogenic abnormalities in either nDNA or mtDNA. Mitochondrial mutations most commonly are inherited (approximately 75%), but can also occur de novo (approximately 25%) [27]. There are more than 250 different pathogenic mtDNA variants, and, in the case of primary mitochondrial myopathies (PMM), they have been discovered in all 37 mtDNA genes [16, 21]. As such, one of the main challenges in the molecular diagnosis of mitochondrial diseases is to distinguish rare or novel causative mutations from nonpathogenic gene polymorphisms. Mitochondrial respiratory chain disorders can be caused by mtDNA mutations in almost 20% of cases, and the rest are caused by mutations in nDNA [27, 30]. The most common cause of mtDNA alterations are point mutations—they are found in 1 of every 200 newborns; however, most individuals remain asymptomatic due to carrying a mutated gene with low heteroplasmy level [27, 31]. In contrast, large-scale mtDNA deletions are rare (1.5 in 100 000)—they most commonly arise as de novo mutations and carry a less than 10% risk of transmission [27].
The m.3260A > G transition in the MT-TL1 gene is an infrequently occurring point mutation, that has been observed to cause maternal myopathy and cardiomyopathy [32, 33], in particular—hypertrophic and dilated cardiomyopathy [21, 32]. It is also related to MELAS syndrome [32, 34]. Hypertrophic cardiomyopathy is the most common form in all patterns of mitochondrial disease, occurring in up to 40% of patients [21, 26], including the patient described in this paper. The absence of mtDNA specific cardiovascular phenotype poses yet another diagnostics challenge to the clinicians. In addition, specific variants can lead to various phenotypes, while similar phenotypes can stem from multiple different mtDNA mutations [21]. However, while cardiac involvement in patients with mitochondrial respiratory chain diseases is common, and the presence of cardiomyopathy drastically decreases the overall survival rate [17, 29, 35], the incidence of severe cardiovascular morbidities is rare, and the prognosis is usually favorable [17].
In addition to myopathy and cardiomyopathy, our patient showed signs of encephalopathy and had eventually developed seizures, subsequently being diagnosed with mitochondrial epilepsy. To our knowledge, mitochondrial epilepsy, caused by m.3260A > G transition, has only been reported in two families with MELAS syndrome [34, 36].
Gathering a thorough family history is an essential part of diagnosis. It is helpful in directing the genetic tests, as well as discovering a potential disorder in other family members—whenever possible, maternal transmission of the disease should be recorded over the preceding three generations. However, notably, mitochondrial diseases have a substantial clinical heterogeneity within affected families, which can make it difficult to establish an accurate genotype/phenotype association and is an additional diagnostic challenge [19, 27]. This variety of symptoms and their severity can be explained by the phenomenon of heteroplasmy—the presence of mutated and wild-type mtDNA in the same individual [37]. In addition, different ratios of mutated and wild-type mtDNA can be observed between multiple affected family members, as well as between various tissues in the same organism [19]. The threshold to exhibit clinical symptoms usually varies between mutation loads of 60% and 90% depending on the specific mutation, with higher percentages leading to more severe phenotypes [32, 38, 39]. Similar findings could be observed in our case; however, the patient had a heteroplasmy level of only 50%, while his asymptomatic mother had a level of 25%. Variable heteroplasmy levels of the same mtDNA variant can even lead to phenotypic expression corresponding to different clinical syndromes—for example, the same mutation in MT-ATP6 gene with a mutation load of 60–90% causes the less severe NARP, while a load of more than 90% causes the more clinically severe maternally inherited Leigh syndrome (MILS) [11].
Notably, the heteroplasmy level observed in our patient was low (50%); however, he displayed multiple serious clinical features, including myopathy and epilepsy. This could be explained by the fact that the mtDNA used in analysis was obtained from a blood sample. Analyzing samples from other tissues (for example, muscle, nervous) would most likely have yielded markedly higher levels [34].
Nowadays, the pharmacological treatment of mitochondrial disorders involves using various combinations of vitamins and cofactors that are vital for normal mitochondrial function. The goal of this therapy is to manage the associated symptoms, by promoting key enzymatic reactions, while reducing excess of free radicals and accumulated toxic acyl coenzyme A (CoA) molecules [40].
It is our common practice to start empirical trial of mitochondrial cofactors as soon as the mitochondrial disease is suspected, especially in acute cases. The components of the “cocktail” can be varied, but similarly to the case presented in this paper, we usually include supplements, such as carnitine and thiamine.
Carnitine is a cellular compound that transfers long-chain fatty acids across the mitochondrial inner membrane—a process that is vital for mitochondrial β-oxidation of fatty acids and the esterification of free fatty acids that would otherwise be sequestered by CoA. Its supplementation is used to restore free carnitine levels leading to the removal of accumulating toxic acyl compounds [40]. However, shortly after beginning the therapy, our patient suffered from diarrhea, which is a known side effect of carnitine [40], and, due to the lack of effectiveness, this treatment was temporary halted.
Thiamine is used to augment pyruvate dehydrogenase activity, thus increasing the catabolism of pyruvate to acyl CoA [41]. It has shown a good effect in lowering the blood lactate levels in our previous patients. Similarly to carnitine, and other components of the “cocktail” used to treat this patient, it is also readily available.
After establishing the diagnosis, two more compounds were added: CoQ10 and riboflavin. CoQ10 is a vital component in mitochondrial electron transport chain shuttling electrons from complexes I and II to complex III. Supplementing CoQ10 restores electron flow, thus improving the clinical manifestations of various mitochondrial diseases.
Riboflavin is a flavoprotein precursor, thus, functioning as an important building block in aforementioned complexes I and II. It is effective in treating various mitochondrial diseases, particularly those with complexes I and II deficiencies [40, 41].