
Isala cohort cross-sectional study
The Isala citizen-science project (https://isala.be/en/) was approved by the Ethical Committee of the Antwerp University Hospital/University of Antwerp (B300201942076) on November 18th, 2019, and registered online at clinicaltrials.gov with the unique identifier NCT04319536. A detailed study design and cohort information can be found in Lebeer et al. [8].
GeneDoe study design and communication
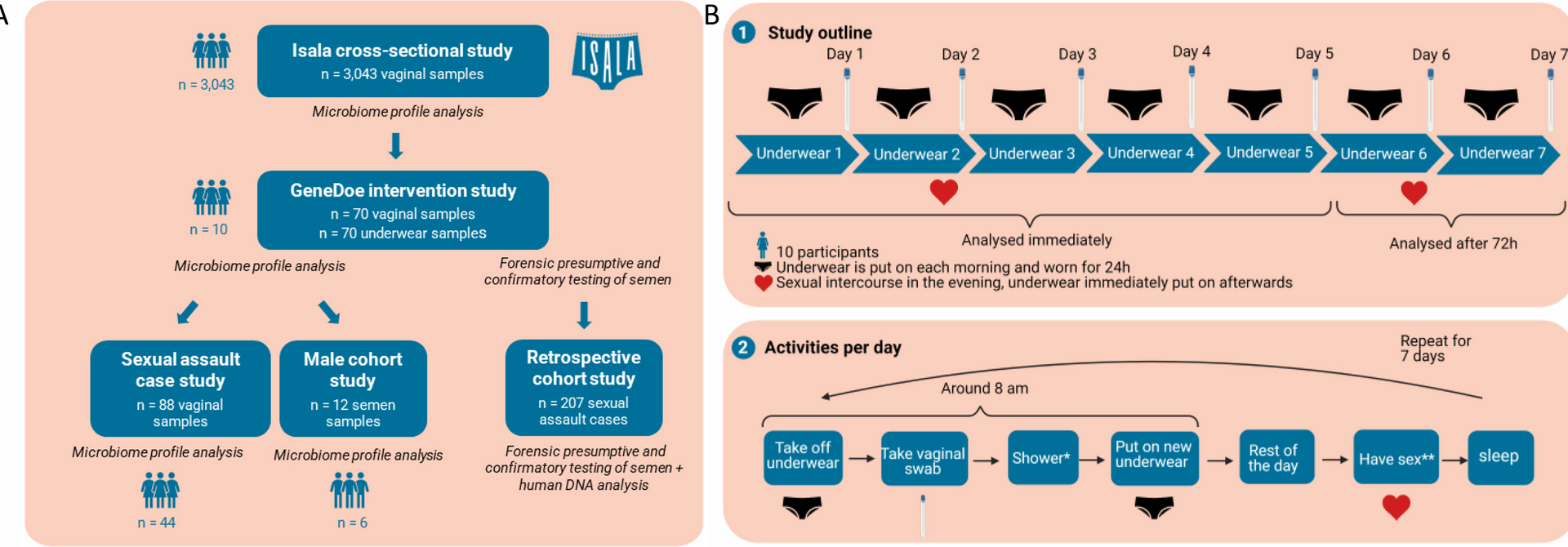
Ten self-reported healthy, premenopausal women were recruited. Signed informed consent was obtained from all participants. This study was approved by the Ethical Committee of the Antwerp University Hospital/University of Antwerp (B3002021000066) on March 23rd, 2021. We aimed for an overlap in their menstrual cycles, allowing sampling on 8 consecutive days without any of the participants menstruating. To ensure that the study was carried out as optimally as possible, clear communication with our participants was essential for which mailings were used as the primary tool. Participants were first given some basic background information framing the study together with a first short summary of the goal of the study and what was expected from them. When participation was confirmed, communication proceeded via e-mail to obtain information about sizes for underwear, among other things. To ensure optimal procedures during the sampling days, a custom, creative brochure was designed. These brochures contained all the information with regard to optimal sampling. Participants were introduced to the term “swab” and guided through the self-swabbing procedure in a step-by-step, detailed protocol. Aside from the swabbing instructions, each participant provided a clear overview of which samples needed to be taken each day and of when and how delivery at our laboratory would occur.
Sample collection with self-sampling kits
Ten participants were provided with a self-sampling kit containing seven pieces of underwear (Marks & Spencer London, 100% cotton, identical for every participant and washed uniformly as described below) and seven FLOQSwabs® in a sterile, dry 15-ml tube. Each day, participants were asked to (1) wear the (provided) underwear for 24 h; (2) take off the underwear of the day before; (3) put it in the provided paper bags; (4) fill in the paper bag label concerning the time of sexual intercourse, use of a condom, lubricant use, and partner’s ejaculation; (5) take a vaginal swab each morning; (6) take a shower if preferred; and (7) change into newly provided underwear for the next 24 h. On particular days, they were asked to have no sexual intercourse, while for two specific days (evenings of days 1 and 5), they were asked to have sexual intercourse with their partner before sleeping. On evenings of sexual intercourse, the participants were asked to put their underwear back on immediately afterwards and not to go to the toilet for 1 h; if they preferred, they could use a dry wipe. After each underwear change, the participants delivered their worn underwear packed in a paper bag, together with the swab of that morning, to the lab for immediate processing.
Textile substrate
The underwear was washed before use. A standard washing program at 30 °C together with standard washing detergent and softener was used, after which the samples were air-dried for 24 h on disinfected drying racks in a room separated from any passage. Once dry, every pair of underwear was put separately in a bag. This whole process was performed using protective gear such as gloves and a mouth mask to avoid contamination.
DNA extractions for the intervention study
Underwear was cut according to the seams of the vaginal-oriented part of the underwear (approximately 165 cm2). This piece was placed in a 50-ml tube with 15 ml of PBS (1x) using sterile tweezers. The tubes were shaken for 10 min (MO BIO Vortex Genie® 2 Vortex) before 10 min of centrifugation at 4 °C and 9000 rpm. Sterile tweezers were again used to wash every piece for maximum retrieval. Five hundred microliters of the remaining solution was then used for DNA extraction. The FLOQSwabs® were prepared for DNA extraction with the addition of 1.5 ml of CD1 solution to the swab. The swabs were incubated for 15 min at room temperature before being vortexed thoroughly for 15 s each. A total of 1.3 ml was then used for DNA extraction.
DNA was extracted using the DNeasy PowerSoil Pro Kit (Qiagen). The extraction itself was performed following the manufacturer’s manual, with adjustments to a starting volume of 500 µl and a 50 µl elution volume. For elution, 50 µL of C6 solution was added to the MB spin column and incubated for 5 min at room temperature. After the incubation, the spin column was centrifuged for 1 min at 15,000 × g. The flowthrough was subsequently added to the spin column and incubated for 3 min at room temperature before being centrifuged for 1 min at 15,000 × g. Afterwards, the extracted DNA was quantified via spectrophotometry (Take 3, BioTek) and fluorometry (Qubit 3.0 fluorometer, Life Technologies) and later stored at − 20°C.
Sample selection of sexual assault kits
On 2 September 2019, ethical approval was obtained from the central ethical committee UAntwerp/UZA under EDGE455 to analyze Sexual Assault Evidence Collection Kits (SAECKs) from the forensic DNA lab of Antwerp University Hospital. Between 2003 and 2021, 1350 SAECKs were collected from sexual assault victims in the juridical district of Antwerp. By briefly reading the reports, a selection of SAECKs with the highest relevance was made for this study. We included only studies containing DNA extracts (by differential lysis with a QIAamp® DNA Mini Kit) for which both vaginal swabs and underwear extracts were available (n = 88). Finally, we selected SAECKs with the aim of obtaining an even distribution of DNA extracts over the years (i.e., a maximum of 5 SAECKs per year) (n = 44). Together, these findings led to the inclusion of total DNA extracts (the S and V fractions) in the form of vaginal swabs (Nswabs = 44, Nextracts = 88). All DNA extracts were stored at the Forensics DNA Laboratory of University Hospital Antwerp in 1.5-mL tubes at − 20°C. After searching for the selected SAECKs with their DNA extracts, the samples were thawed and vortexed. Finally, 20 μL was transferred to a new 1.5-mL tube for 16S rRNA sequencing. Given that these samples underwent standard forensic analyses with a focus on human DNA, they included both a potential victim DNA fraction (non-sperm fraction) and a potential suspect DNA fraction (sperm fraction) sample for each vaginal sample. Since the microbial compositions of both DNA fractions were similar, we pooled both fractions (Supplementary Figure S5).
16S rRNA amplicon sequencing
Amplification via PCR was performed for all samples, including negative controls (i.e., molecular-grade water and DNA extraction controls), using 96-well plates as described in Ahannach et al. [58]. According to the biomass of the samples, based on DNA quantification, different compositions of mastermix (MM) were prepared. Two-microliter samples with high biomasses were added to 14 µl of MM and 4 µl of 2.5 mM barcoded forward and reverse primer mix (2 µl forward and 2 µl reverse) in a single well. For the low-biomass samples, 5 µl was added to 11 µl of MM, and 4 µl of 2.5 µM barcoded forward and reverse primer mix was added (2 µl forward and 2 µl reverse). The primers used in this PCR were the 515F-806R primers, which were altered for dual-index paired-end sequencing, as described in Kozich et al. [61]. The sealed plates were centrifuged shortly before the PCR program was started. A 1% agarose gel was used to verify the resulting PCR products. The gel was loaded in each well with 1 µl of PCR product and 5 µl of loading dye (1:10) using a GeneRuler 1Kb Plus ladder (Thermo Scientific™). The gel was run for 30 min at 100 V to determine if the amplification was successful. Next, the PCR products were purified using Ampure XP (Agencourt) with a magnetic block following the manufacturer’s protocol. After purification, the DNA concentration was again measured using a Qubit 3.0 fluorometer, and the library was prepared by pooling equimolar concentrations of all the samples. Thirty microliters of the library was loaded with 6 µl of loading dye on a 0.8% agarose gel, together with a GeneRuler 1Kb Plus ladder. After running for 50 min at 60 V, gel extraction was performed using the NucleoSpin Gel and PCR Clean-up Kit (Macherey–Nagel) according to the manufacturer’s protocol. The final elution step of this kit was adjusted, whereas the DNA was eluted using 15 µl of the provided elution buffer and subsequently reloaded on the column for increased yield. The concentration was measured using a Qubit 3.0 fluorometer to dilute the library to 2 nM, which was again confirmed with a final Qubit 3.0 fluorometer. Next, 0.2 N NaOH (Illumina, San Diego, California, USA) was used for denaturing the library, which was subsequently diluted to 6 pM and spiked with 10% Phix control DNA (Illumina). The V4 region of the 16S rRNA gene was sequenced using dual-index paired-end sequencing (2 × 250) with a MiSeq Desktop sequencer (Illumina).
Forensic presumptive and confirmatory testing of semen via underwear and vaginal swabs
First, pictures were taken from each piece of underwear, and vaginal secretion amounts were subjectively described (i.e., little or average). Next, an alternative light source (Crime-Lite® 2, 420–470 nm BLEU, Foster & Freeman) was used to detect the fluorescence of possible semen stains. Detection with Crime-lite served as an indication to cut a small piece (approximately 0.5 cm2) of the underwear and the tip of the vaginal swab for PSA testing (PSA-CHECK-1, Veda-Lab). Then, 300 µl of 0.9% NaCl was added to the the piece of underwear, which were vortexed thoroughly and incubated for 10 min at room temperature. After incubation, 225 µl was added to the sample well, and the results were interpreted after 10 min. In case of suspicion of the presence of blood due to reddish spots after visual inspection with natural light and Crime-light®, an additional blood test (BLUESTAR® OBTI) was performed on the underwear. A small piece (approximately 0.5 cm2) of underwear was added to 20 µl of Tris buffer (pH 7.8), vortexed thoroughly and incubated for 5 min at room temperature. Then, 120 µl was added to the sample well, and the results were obtained after 5 min of incubation.
Semen sample collection and processing dataset
The FORmics project at the Institute of Forensic Medicine in Switzerland was approved by CEBES, the Ethics Review Board at the University of Zurich, and received the following identifier 2021-11c. Semen samples were self-collected by 6 male participants. For each of these, two biological replicates were generated by depositing 50 μL of sample on a swab, resulting in a total of 12 samples. DNA was extracted with the QIAamp BiOstic Bacteremia DNA Kit (Qiagen), and library preparation for the V3–V5 16S rRNA gene region was carried out with the F357 and R926 primers (detailed in Table S4). The volume of the library preparation PCR mixture was 25 μL, and the mixture contained 2X Phusion Hot Start II High-Fidelity PCR Master Mix. The reaction mixture contained 1 μL of DNA, 12.5 μL of 2X Phusion Hot Start II High-Fidelity PCR Master Mix, 1.25 μL of forward primer, 1.25 μL of reverse primer, and 9 μL of PCR-grade water. A PCR negative control was included where the reaction well contained 1 μL of PCR-grade water. The thermocycling conditions for the amplicon PCR were as follows: 30 s at 98 °C for the initial incubation followed by denaturation; 32 cycles of 10 s at 98 °C and annealing for 30 s at 50 °C for the V3–V5 region; extension for 25 s at 72 °C; a final extension for 10 min at 72 °C; and hold at 4°C. The integrity of the amplicon PCR products was checked using 1.5% agarose gel electrophoresis, where the gel was loaded with 3 μL of PCR product, 2.5 μL of distilled water, and 3 μL of dye. The PCR products were purified using magnetic beads according to the manufacturer’s protocol (AMPure XP, Beckman Coulter) at a ratio of 0.8-fold the volume of AMPure XP beads per volume of PCR product. Next, the purified amplicon DNA was dual-indexed using the Nextera XT Index Kit. PCR was performed in a total volume of 25 μL with 5 μL of purified PCR product, 12.5 μL of 2X Phusion Hot Start II High-Fidelity PCR Master Mix, 2.5 μL of Index 1, 2.5 μL of Index 2, and 2.5 μL of molecular-grade water. The thermocycling conditions for the index PCR were 30 s at 98 °C for initial incubation, followed by 12 cycles of denaturation for 10 s at 98 °C, annealing for 30 s at 55 °C, extension for 20 s at 72 °C, a final extension for 5 min at 72 °C and holding at 4°C. The indexed PCR products were purified with AMPure XP beads at a ratio of 0.6-fold the volume of AMPure XP beads per volume of PCR product. The library products were quantified using a Spark 10 M Multimode Microplate Reader (Tecan) and a Qubit dsDNA BR assay (Thermo Scientific). Libraries were normalized to 3.03 nM, pooled, denatured with 0.2 M NaOH, and diluted to a final concentration of 20 pM. A PhiX sequencing control (Illumina) was used for sequencing. The PhiX library was prepared by diluting the 10 nM PhiX library (2 µl) with 10 nM Tris–Cl, pH 8.5 (3 µl), to 4 nM. Then, 5 µl of the 4 nM PhiX library was denatured with 5 µl of 0.2 N NaOH. The denatured PhiX was diluted to 20 pM by adding 990 µl of prechilled HT1 solution to 10 µl of the PhiX library. Sequencing was carried out on an Illumina MiSeq platform (Illumina, Inc., Hayward CA, USA) with the MiSeq Reagent Kit V3, generating paired-end reads of 2 × 300 bp. The raw reads were trimmed with Cutadapt v3.5 and quality filtered and denoised with the DADA2 pipeline [version 1.18.0] with the following parameters. (maxN = 0, maxEE = c(4,6), truncQ = 2), resulting in an ASV abundance table for 12 semen samples.
Retrospective study
To study the prevalence of risk factors, medico-legal examinations and SAECK analysis, medicolegal findings were correlated with DNA analysis results, providing novel information that could be used to design guidelines and/or suggestions to optimize the current approach in cases of sexual assault. A retrospective descriptive case study (Local KULeuven ethics committee [mp13636]) of 207 sexual assault cases was performed by the Laboratory of Forensic Genetics [UZ Leuven] from 2012 until 2016. After excluding 56 cases, 151 cases were left for analysis. The exclusion criteria were as follows: a forensic analysis (biological trace evidence examination and DNA profiling) had to have been requested by the court; the case was a sexual assault; and PSA tests for the presence of semen had to be performed. The remaining 151 cases were studied in more depth with regard to background information, time and date, medical examination, and DNA analysis.
Data analysis
The abovementioned results were analyzed with R-studio (version 1.4.1106) using the in-house-developed tidyamplicons package (github.com/Swittouck/tidyamplicons) and the tidyverse package for analyzing diversity between samples (beta diversity) and within samples (alpha diversity). For alpha diversity, Shannon diversity was used as a measure of entropy. The Bray‒Curtis distance was used to measure the beta diversity between samples. With respect to the Isala dataset, we tested the association between reported sexual intercourse in the last 24 h and no intercourse, adjusting for age, recent antibiotic usage, and technical covariates, using six different differential abundance methods: ALDeX2 [62], ANCOM-BC [63], DESeq2 [64], Limma (with voom transformation) [65], a linear model fit on the Centered Log Ratio transformation [66], and Maaslin2 [67]. Each factor was adjusted for technical confounders, including run and library size, and adjusted for age and antibiotic usage in the last 3 months. The Benjamini–Hochberg procedure was used to control the False Discovery Rate and correct p values for multiple testing. In the GeneDoe dataset, we added a random effect per participant to pair samples; as a result, only the Limma, CLR, and Maaslin2 methods could be applied, as the others do not support random effects. For GeneDoe, the reported p values are nominal and were not corrected for multiple testing because the sample size was too small and we lacked power. All these methods were implemented in a unified interface in the multidiffabundance R package (https://github.com/thiesgehrmann/multidiffabundance).
To classify samples into post-coital and non-post-coital samples, we needed to create a balanced dataset. With 410 individuals who had reported intercourse within the last 24 h, we created a balanced dataset by sampling an equal number of individuals who indicated that they had not had intercourse within the last 24 h. To ensure that performance was not based on a fortuitous sampling, we repeated this process 10 times. Within each downsampling, we performed a tenfold cross-validation. In the cross-validation loop, we trained a logistic elastic net classifier with a fixed alpha (penalty weight) of 0.5. An inner cross-validation loop was used for hyperparameter optimization (L1 and L2 mixing ratios). We evaluated the performance of the classifier with respect to the area under the curve (AUC) and determined whether these scores were significantly better than random with a permutation test in which the posterior probabilities were permuted and the area under the curve (AUC) recalculated. Within each fold, we selected the posterior probability (post-coital score) threshold that resulted in the highest F1 score within each fold. For the final model, we used the average threshold across the 10 samplings and the 10 folds of the cross-validation, resulting in a threshold of 0.399. Using the same downsampling sets as before, we trained 10 models on the fully balanced datasets. Finally, we tested these models against the GeneDoe and sexual assault kit datasets, and we report the performance as the average and standard deviation of the predictions across all repeats. The weights reported in supplementary Table S3 reflect the average weights of the full models across the 10 down-samplings. In addition, ASVs from the GeneDoe study (for which only the V4 region was sequenced, 2 × 250 bp) were effectively matched within the broader ASVs (Table S2) from the Swiss seminal microbiome (for which the V3–V5 hypervariable regions were sequenced, 2 × 300 bp) using exact substring matching.