
GNE myopathy was first described by Nonaka as a hereditary disorder. It is commonly manifested by distal muscle weakness with a gradual progression, initially affecting the ankle dorsiflexors and subsequently leading to weakness in the legs, arms, and potentially the cervical muscles. GNE myopathy predominantly manifests in individuals during their third decade of life, with the onset of the condition being rare before the age of 10 or after the age of 50 [6]. In our study, the onset of symptoms occurred at age 17 for Patient 3 and at age 49 for Patient 6. And a cohort study involving 125 Chinese patients with GNE myopathy whose onset age varied from 3 to 60 years, exhibiting the heterogeneity of onset age [6]. Typically, the upper extremities are impacted within 5 to 10 years following disease onset, and loss of ambulation may occur within 10 to 20 years. In the advanced stages, involvement of the neck muscles occurs, resulting in weakened neck flexion [4, 7, 8]. A cohort study indicates that the majority of GNE myopathy patients encounter challenges in ambulation, stair climbing, or experience frequent falls. As the disease progresses, patients exhibit a decline in functional abilities across the domains of mobility, upper extremity function, and self-care. Additionally, certain patients exhibited muscle spasms or pain that were not observed within our study [8]. Moreover, Mori-Yoshimura et al. conducted a cohort study involving 24 patients diagnosed with GNE myopathy. Over the one-year follow-up, the researchers observed disease progression in 19 patients, which was primarily characterized by increased weakness in the hands and neck, gait disturbances, or exacerbated lower limb weakness [7]. The muscle weakness in GNE myopathy is usually symmetrical, and this pattern has been confirmed by Yan et al. [7]. The symmetrical weakness was also observed in our study. In this study, all patients exhibited weakness in the distal lower limbs at disease onset. Different from the typical pattern, Patient 2 experienced atrophy of the interosseous muscles in the first and second phalanges of both hands within three years following disease onset, while Patient 1 demonstrated early onset of neck weakness. The observed discrepancies highlight the heterogeneity of the clinical manifestation of GNE myopathy.
Beevor’s sign, characterized by the upward movement of the umbilicus during neck flexion, indicates weakness in the hip flexors and lower abdominal muscles. This sign is rarely observed in GNE myopathy, and is only reported in the Indian GNE myopathy cohort with a 59.2% prevalence rate in 157 Indian patients [9, 10]. While Beevor’s sign is an indicator of facioscapulohumeral muscular dystrophy, it has also been reported in other diseases such as Pompe disease and myotonic dystrophy. Moreover, Beevor’s sign may serve as a valuable clinical marker for diagnosing GNE myopathy, especially when it is associated with the preservation of the quadriceps femoris muscle [11]. In this study, Patient 5 exhibited Beevor’s sign, which may be attributed to the asymmetric involvement of trunk muscles. This is the first report of Beevor’s in a non-Indian GNE myopathy patient and may indicate an atypical pattern of involvement.
Respiratory dysfunction is a rare occurrence in GNE myopathy. Cohort studies conducted among Iranian non-Jewish Persian and UK populations have documented an absence of respiratory dysfunction cases [12, 13]. However, to East Asian patients, a long disease duration seems to be a risk factor for respiratory dysfunction. Patient 5, who has experienced a disease duration of 37 years, demonstrated a reduced %FVC (76.3%). Similarly, a Japanese GNE myopathy patient with a 43-year disease duration exhibited a low %FVC (67.5%), despite maintaining diaphragm function [14]. Both two patients exhibit trunk muscle involvement, which suggests involvement of trunk muscles may contribute to respiratory impairment. A Japanese cohort study analyzed respiratory function test data from 56 patients and identified reduced FVC in 7 individuals, whose disease duration spanned 17 to 36 years [15]. Moreover, A one-year follow-up study involving 24 GNE myopathy patients reported a significantly decreased FVC as the disease advances, with more pronounced declines observed in non-ambulatory patients [7]. These findings indicate that respiratory dysfunction is associated with long disease duration and highlight the need for clinicians to monitor respiratory function in patients with GNE myopathy, especially those who are non-ambulatory.
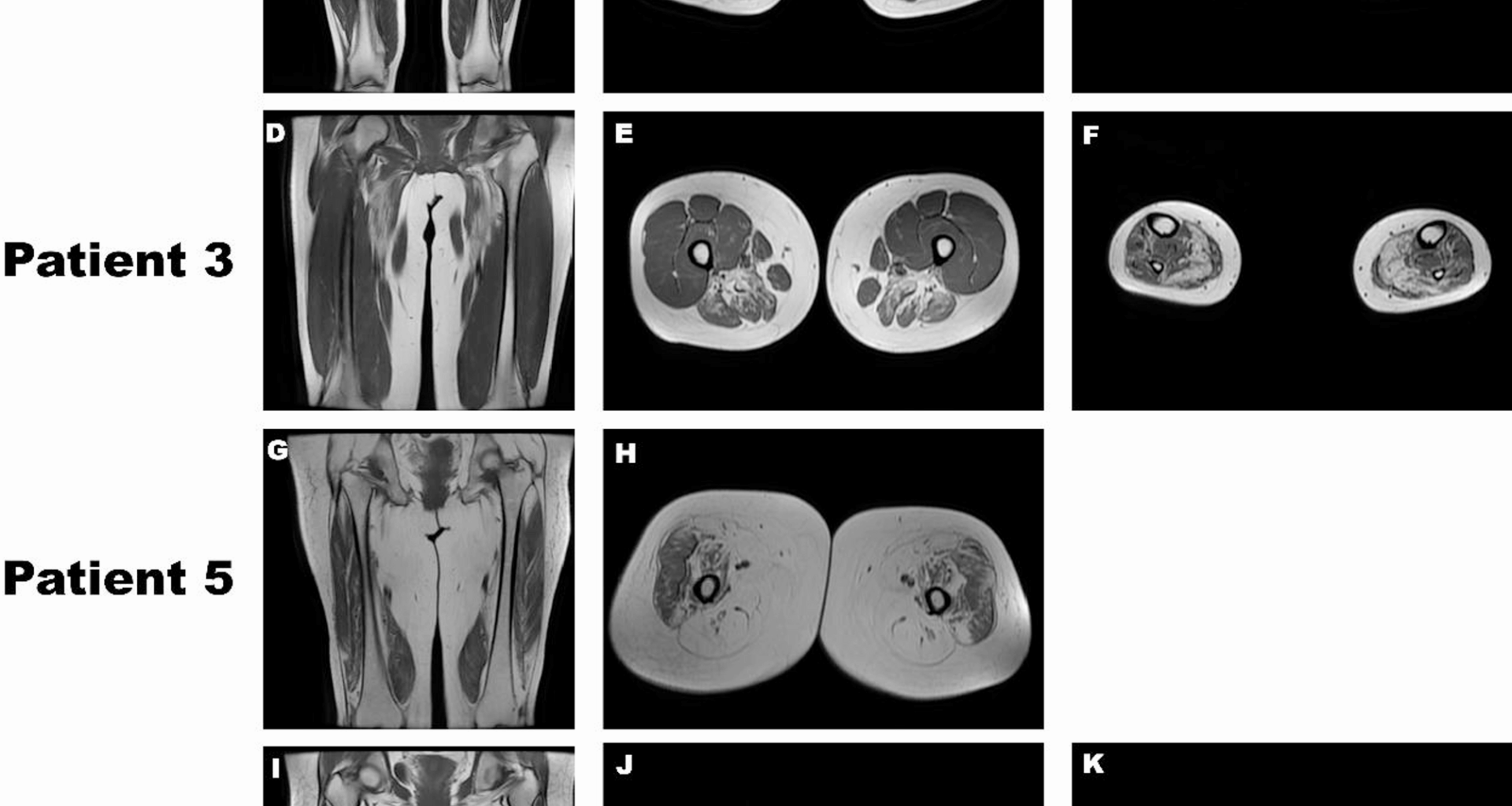
Muscle MRI revealed involvement of the anterior compartment of the lower legs, in addition to the posterior and medial compartments of the thigh. During the initial stages of the disease, the soleus and gastrocnemius medialis are implicated. Furthermore, a study investigating the imaging characteristics of rimmed vacuolar myopathies revealed heterogeneity in the affected patterns of thigh and lower leg muscles in GNE myopathy [16]. Shahriar Nafissi et al. divided the muscles of the lower leg and thigh into three clusters according to the degree of involvement, in which adductor magnus, tibialis anterior, semitendinosus, and semimembranosus muscles exhibited the most severe involvement [17].In this disease, the quadriceps femoris was the final muscle to be affected, with the rectus femoris being involved first and the vastus lateralis being the last [18]. In this study, MRI scans of the muscles in Patients 1, 3, and 6 revealed significant involvement of the posterior thigh muscles. Conversely, the lower-leg muscle involvement was relatively mild compared to the typical pattern, particularly in Patient 6. A cohort study of Russian GNE myopathy patients identified a similar pattern in one individual, whose MRI demonstrated significant involvement of the posterior thigh muscles, with relative sparing of the lower-leg muscles, except for the soleus [19]. Additionally, Patient 3 exhibited mild involvement of the tibialis anterior muscle, which is atypical for GNE myopathy. In Patient 5, the thigh muscles were nearly completely atrophied, with the quadriceps femoris also affected, particularly the rectus femoris, whereas the vastus lateralis was the least affected. Muscle MRI provided useful, noninvasive tools to evaluate patients with GNE myopathy. Nuria Carrillo established an MRI grading system that correlates with the motor function, categorizing GNE myopathy into four distinct stages. Identifying the MRI stage in GNE myopathy is beneficial to assess prognosis and inform treatment strategies. Neurologists should particularly focus on patients in stages 2 and 3 to prevent progression to stage 4 [18]. The muscle involvement in GNE myopathy exhibits distinct features and heterogeneity in imaging and underscores the need for further investigation into the diverse patterns of muscle involvement in GNE myopathy for better prediction of patient prognosis.
Previous research has demonstrated that the cell membrane restricts the release of substantial quantities of CK into the bloodstream, resulting in mild to moderate increases in serum CK levels in the majority of patients. Nevertheless, the normal or mild increases in levels of CK might also be attributed to the substantial degradation of muscle cells [9, 20]. Patient 5 exhibited the most severe clinical manifestations in the study, with muscle MRI revealing extensive muscle atrophy. However, her CK level remained within normal limits. This may reflect advanced skeletal muscle damage.
Similar to other myopathies, the neurophysiology of GNE myopathy is characterized by a myopathic pattern. Typical myopathic pattern has been observed in the majority of GNE myopathy patients across various cohorts. However, certain patients displayed a mixed pattern, while a few demonstrated a neurogenic pattern [19, 21, 22]. The previous study demonstrated that characteristics of axonal neuropathy, including reduced CMAP amplitude and the presence of large-amplitude, long-duration motor unit action potentials (MUAPs), are observable in GNE myopathy. however, the underlying cause of this phenomenon remains unclear. Initially, the neurogenic pattern was considered as pseudoneurogenic pattern due to alterations in muscle fibers associated with chronic myopathies. Nevertheless, the hyposialylation of gangliosides in motor nerve axons may contribute to axonal neuropathy in GNE myopathy, and the neurogenic pattern was also observed in the unaffected muscle, suggesting a lesion of the peripheral nerve [23, 24]. The neurophysiological patterns associated with GNE myopathy may lead to diagnostic inaccuracies. In the study involving Russian GNE myopathy patients, four patients were initially misdiagnosed with spinal muscular atrophy due to the presence of neurogenic patterns observed in EMG. Furthermore, two of the patients underwent a repeated EMG on the clinical affected muscles, which revealed a myogenic pattern. It is noteworthy that the vastus lateralis was initially selected for EMG in all four patients [19]. Due to relatively sparing in GNE myopathy, the vastus lateralis is more likely to exhibit neurogenic rather than myogenic damage, which suggests that EMG should be conducted on clinically affected muscles for more accurate assessment. However, prolonged disease duration and severe atrophy may also result in a pseudoneurogenic pattern, which highlights the importance of comprehensive clinical assessment of GNE myopathy. This study identified a reduction in CMAP amplitude in the peroneal nerve among the majority of patients, aligning with previous study. Although the most patients exhibited a myopathic pattern, some patients also demonstrated a mixed pattern or neurogenic pattern. Specifically, Patient 2 exhibited a neurogenic pattern in the right tibialis anterior, indicating a potential peripheral nerve lesion. In addition, Patient 5 displayed a suspected neurogenic pattern, however, this may a pseudoneurogenic pattern due to the prolonged disease duration and significant muscle atrophy.
The prevalence of founder or recurrent genetic variations is relatively higher across diverse racial populations. In Japan, the p.V603L and p.D207V variants are the most prevalent, while the p.M743T variant is a hotspot in the Middle East. The p.I618T variant is frequently reported in Bulgaria [4, 9]. The p.D207V variant is known to be the hotspot variant in the Chinese patients and we identified the p.D207V variant in three patients in the study. It has been proposed that a correlation may exist between genotype and phenotype in GNE myopathy [9]. A previous study categorized Chinese GNE myopathy patients into two groups based on the presence or absence of the p.D207V variant. The study demonstrated that individuals without the p.D207V variant experienced an earlier onset of the disease, lost the ability to walk independently at a younger age, and had an increased risk of becoming wheelchair-dependent [25]. Likewise, a study analyzing the clinical manifestations between patients with the p.D207V/p.V603L compound heterozygous variant and those with the p.V603L homozygous variant in Japanese patients found that patients with the p.D207V/p.V603L compound heterozygous variant exhibited milder clinical features, such as the later age of onset, preserved respiratory function, maintained independent walking ability, and higher grip strength [26]. These observations suggest that the p.D207V variant is correlated with a milder disease phenotype. Due to the extreme rarity of the homozygous p.D207V variant, it remains unclear whether there is a phenotypic difference between the homozygous p.D207V variant and the heterozygous p.D207V variant. Yan et al. found patients with homozygous p.D207V had an earlier onset age than compound heterozygous ones [6]. Besides, Zhu et al. reported a patient with the homozygous p.D207V variant who presented with mild clinical manifestations. This observation suggests that the homozygous p.D207V variant may present with exceptionally mild symptoms, potentially resulting in underdiagnosis [27]. Furthermore, five novel variation sites were discovered in our study, which broaden the mutational spectrum.
RVs are the key histopathological maker of GNE myopathy, which were consistently observed in all anterior tibial muscle biopsies in the study conducted by Yan et al., highlighting the significance of selecting an appropriate biopsy site [6]. In our study, all muscle biopsies were performed from the biceps brachii or gastrocnemius, which may account for the absence of RVs in one patient. RRFs serve as a crucial biomarker for mitochondrial damage. In the present study, RRFs were found in two patients, suggesting mitochondrial dysfunction in individuals with GNE myopathy. The pathogenic variant in the GNE gene compromise mitochondrial structure and function, induce mitochondrial transmembrane depolarization, and consequently initiate apoptosis in cell mode was confirmed by Ranjana Arya et al. [28]. Furthermore, an analysis of genomic expression profiles in skeletal muscle from GNE myopathy patients and healthy individuals demonstrated that differential expression genes linked to mitochondria constituted 18.6% of the overall differences, including mitochondrial function, structural organization, and the encoding of transport proteins. Importantly, aberrant mitochondrial morphology was also observed in the skeletal muscle cells of patients with GNE myopathy [29]. These findings indicate that mitochondrial dysfunction may represent a critical event in the progression of GNE myopathy. Inflammatory cell infiltration is not typically observed as a pathological feature of GNE myopathy. Nonetheless, increased ACP activity was identified in Patient 2, indicating possible inflammation. Hanns Lochmuller reported that an Iranian non-Jewish GNE myopathy patient presented with muscle inflammation and the extensive infiltration of inflammatory cells, indicating the presence of inflammatory responses in skeletal muscle should not preclude the diagnosis of GNE myopathy [30]. However, extensive infiltration of inflammatory cells was not observed in Patient 2. Consequently, the increased activity of ACP may be attributed to secondary cellular damage.
Recent advancements in the treatment of GNE myopathy have focused on sialic acid supplementation. Satoru Noguchi et al. revealed supplementation with ManNAc, NeuAc, or sialyllactose could significantly mitigate skeletal muscle atrophy, preserve motor function, and prevent myofiber degeneration in mouse models of GNE myopathy [31]. In a subsequent study, the team observed that 6’-sialyllactose exhibited a dose-dependent response. The high-dose group showed greater efficacy in improving motor function, muscle strength, and skeletal muscle pathology in mouse models [32]. These experiments have validated the treatability of GNE myopathy and indicated that administering high doses of sialic acid to enhance the sialylation of skeletal muscles constitutes an effective therapeutic approach for this condition. Clinical trials conducted with Japanese patients have shown that oral aceneuramic acid can maintain or delay the degeneration of muscle strength in GNE myopathy patients, showing both therapeutic efficacy and acceptable safety profiles [33]. After conducting extensive research, Japan approved an extended-release formulation of aceneuramic acid in March 2024 for the treatment of GNE myopathy [34]. The clinical implementation of sialic acid supplementation therapy has markedly increased the significance of screening for GNE myopathy.
This study presents several limitations. Firstly, despite GNE myopathy being a rare condition, the cohort size is relatively small, potentially restricting the generalizability of the results. Secondly, due to its cross-sectional design, this study does not include longitudinal follow-up data on disease progression. Thirdly, pedigree verification was not performed for five patients (Patient 1, 2, 4, 5, and 6), and consequently, the compound-heterozygous status of the variants in these individuals remains unconfirmed.