
Source of extended spectrum beta-lactamase-producing Klebsiella pneumoniae species complex isolates
A total of 24 ESBL-KpSC isolates were analyzed. Of these, 15 (62.5%) were obtained from orthopedic patients with open fractures, four (16.7%) from non-medical caretakers of orthopedic patients, four (16.7%) from the immediate hospital environment (including a sink in a common bathroom, a bed in the ward, the ward floor, and a wheelchair), and one (4.2%) isolate was collected from a healthcare worker (Table 1).
Table 1 Demographic characteristics of 24 extended spectrum beta-lactamase-producing Klebsiella pneumoniae species complex isolatesSpecies and lineages of extended spectrum beta-lactamase-producing Klebsiella pneumoniae species complex isolates
Of the 24 ESBL-KpSC isolates analyzed using Kleborate taxonomic criteria, three Klebsiella species were identified: Klebsiella pneumoniae [14 isolates (58.3%)], Klebsiella quasipneumoniae subsp. similipneumoniae [seven isolates (29.2%)], and Klebsiella variicola subsp. variicola [three isolates (12.5%)]. The 24 ESBL-KpSC genomes were grouped into 19 sequence types (STs), reflecting a high genetic diversity of 79.2% (19/24). Among the 19 STs, two genomes were represented in each of the following STs: ST17, ST307, ST2478, ST367-2LV, and ST3946-1LV, while the remaining genomes were singletons. (Table 1; Fig. 1).
Fig. 1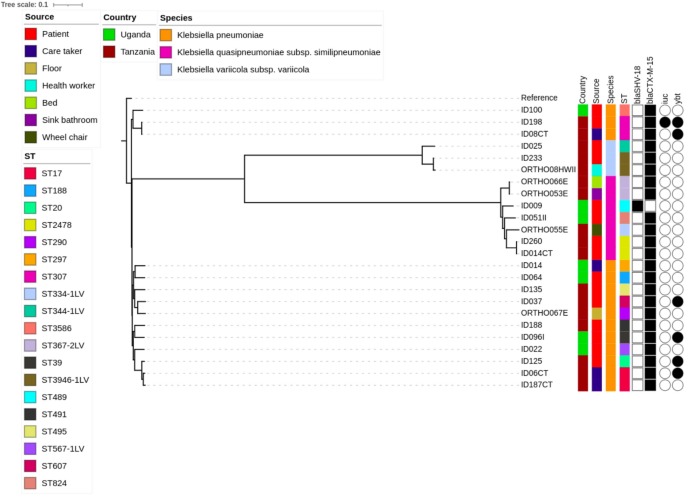
A phylogenetic analysis was conducted to demonstrate the diversity of ESBL-producing Klebsiella pneumoniae species complex genomes from Uganda and Tanzania. The resulting phylogenetic tree highlights the significant diversity of the isolates analyzed in this study, showing multiple sequence types (STs). This wide diversity indicates a lack of a clear clonal pattern, even at the country or hospital level
Predicted capsular polysaccharide and lipopolysaccharide serotypes or loci of extended spectrum beta-lactamase-producing Klebsiella pneumoniae species complex isolates
Among the 24 ESBL-KpSC genomes analyzed, K (Capsular polysaccharide) loci were successfully predicted in 21 out of 24 (87.5%), while 3 genomes (12.5%) had Kaptive K loci confidence calls of ‘Low’ or ‘None,’ indicating fragmented K locus assemblies (i.e., the K locus was not fully assembled in a single contig). Of the 21 genomes with successfully predicted K loci, 16 different K serotypes/loci were detected. Three genomes (14.3%) had the KL102 locus, and two genomes (9.5%) were assigned to each of the following K serotypes: K12, K25, K28, and KL133. The remaining serotypes/loci were each represented by a single genome (Table 1).
Similarly, O (Lipopolysaccharide) loci were successfully predicted in 20 out of 24 genomes (83.3%), while 4 genomes (16.7%) had Kaptive O loci confidence calls of ‘Low’ or ‘None,’ indicating fragmented O locus assemblies (i.e., the O locus was not found in one contig and/or gene deletions occurred). Among the 20 genomes with successfully predicted O loci, 8 different O serotypes/loci were identified. Seven genomes (35.0%) were O1 serotype, four genomes (20.0%) were O2afg serotype, and two genomes (10.0%) each corresponded to the O2a, O3/O3a, and OL103 loci. The remaining loci were each represented by a single genome (Table 1).
Virulence genes and scores of extended spectrum beta-lactamase-producing Klebsiella pneumoniae species complex isolates
Of the 24 ESBL-KpSC isolates, six (25.0%) had a virulence score greater than 0, with five genomes (20.8%) scoring 1 and one genome (4.2%) scoring 4. Virulence score of 0 was assigned to 18 (75.0%) genomes. The identified virulence genes encoded for the siderophores yersiniabactin (ybt) and aerobactin (iuc). None of the genomes contained genes coding for the genotoxin colibactin (clb), salmochelin (iro), or hypermucoviscosity-associated genes (rmpADC and rmpA2) (Table 1; Fig. 1).
Antimicrobial resistance genes and scores of extended spectrum beta-lactamase-producing Klebsiella pneumoniae species complex isolates
All 24 ESBL-KpSC isolates analyzed had an AMR score of 1, as none of them harbored resistance genes coding for carbapenemases or conferring resistance to colistin. The most common ESBL gene identified was blaCTX−M−15, present in 95.8% (23/24) of the isolates, followed by blaSHV−18, found in 4.2% (1/24). The number of drug resistance classes per genome ranged from two to nine, with a median of seven [IQR: 7–8]. The number of AMR genes per genome varied from two to 15, with a median of 10 [IQR: 7–12].
Apart from ESBL genes, the most frequently identified AMR genes encoded resistance to beta-lactams (blaTEM−1D 79.2% [19/24]), sulfonamides (sul2 75.0% [18/24] and sul1 58.3% [14/24]), quinolones (qnrS1 58.3% [14/24]), and aminoglycosides (strB 66.7% [16/24], strA 50.0% [12/24], acc(6’)-Ib-cr 45.8% [11/24], and aac(3)-IIa 45.8% [11/24]). Other notable resistance genes included those for tetracyclines (tet(A) 45.8% [11/24]), phenicols (catII.2 45.8% [11/24]), and trimethoprims (dfrA14 45.8% [11/24]) (Tables 1, 2 and 3; Fig. 1).
Table 2 Distribution of genetic variants of beta-lactamase among 24 extended spectrum beta-lactamase-producing Klebsiella pneumoniae species complex isolatesTable 3 Distribution of antimicrobial resistance genes among 24 extended spectrum beta-lactamase-producing Klebsiella pneumoniae species complex isolatesConvergence of antimicrobial resistance-virulence of extended spectrum beta-lactamase-producing Klebsiella pneumoniae species complex isolates
Of the 24 ESBL-KpSC genomes analyzed, one ST307 isolate exhibited MDR-hypervirulence convergence, defined as genomes with a virulence score ≥ 3 and an AMR score ≥ 1 [14]. This strain, isolated from a patient in Tanzania, was identified as K. pneumoniae harboring the K (capsular polysaccharide) locus KL102 and O (lipopolysaccharide) serotype O2afg. Additionally, the strain contained 13 AMR genes (blaCTX−M−15, AmpC1, blaOXA−1, blaTEM−1D, aac(3)-IIa, aac(6’)-Ib-cr, strA, strB, qnrB1, sul1, sul2, tet(A), and dfrA14), conferring resistance to seven classes of antimicrobials (Table 1).
Plasmid replicon profiles of extended spectrum beta-lactamase-producing Klebsiella pneumoniae species complex isolates
In all 24 ESBL-KpSC isolates analyzed, we used the ABRicate tool with the PlasmidFinder database and identified a total of 14 different plasmid replicon types. The isolates contained between one and five plasmids each, with the most common plasmids being IncFIB(K) present in 87.5% (21/24) of the isolates, followed by IncR in 41.7% (10/24), IncY in 29.2% (7/24), and IncFIB(Mar) in 29.2% (7/24) (Tables 1 and 4).
Table 4 Distribution of plasmid replicons among extended spectrum beta-lactamase-producing Klebsiella pneumoniae species complex isolatesPhylogenetic relatedness of extended spectrum beta-lactamase-producing Klebsiella pneumoniae species complex isolates from Uganda and Tanzania
Our phylogenetic analysis of the 24 ESBL-KpSC genomes from Uganda and Tanzania revealed a heterogeneous tree, indicating a wide diversity of sequence types (STs) among the isolates. The analysis depicted limited clonal patterns, even at the country and hospital levels, further emphasizing the genetic diversity of the isolates studied (Fig. 1).
Interpretation of plasmid mash results from extended spectrum beta-lactamase-producing Klebsiella pneumoniae species complex isolates
Mash distance analysis of Klebsiella pneumoniae clinical isolates revealed distinct diversity patterns across plasmid replicon types, reflecting varying degrees of conservation and evolutionary divergence. IncR plasmids exhibited moderate genetic diversity, with most pairwise Mash distances ranging between 0.072 and 0.102. This range suggests substantial sequence variability among the IncR plasmids, indicative of divergent evolutionary paths. However, select isolate pairs—such as ID014 with ORTHO053E and ORTHO066E—demonstrated lower Mash distances (0.033–0.043), suggesting higher sequence similarity and potentially shared evolutionary ancestry. These results highlight a heterogeneous IncR plasmid population, potentially harboring diverse antimicrobial resistance determinants shaped by distinct recombination and selection events.
IncY plasmids, in contrast, displayed complete sequence identity across all comparisons (Mash distance = 0, 745/745 shared k-mers), signifying the presence of a single, highly conserved IncY plasmid. This uniformity points to clonal dissemination and suggests a stable plasmid backbone that may facilitate the persistent spread of resistance or virulence genes in this clinical setting.
IncFIB(Mar) plasmids were also highly conserved. Five out of six pairwise comparisons showed zero Mash distance, consistent with identical plasmid sequences and supporting recent clonal transmission. A single comparison (ID022 vs. ORTHO055E) yielded a Mash distance of 0.0074, reflecting near-identity and suggesting minimal variation likely due to small-scale structural changes. These findings indicate a clonally circulating IncFIB(Mar) plasmid with potential implications for the dissemination of conserved resistance loci.
IncFIB(K) plasmids revealed a bimodal pattern of diversity. The majority of comparisons—such as ID009 vs. ID051II, ID135, and ID198—produced low Mash distances (0.0045–0.0216), indicative of highly similar or nearly identical sequences, likely resulting from clonal expansion or recent horizontal gene transfer. In contrast, other comparisons involving isolates like ID014CT, ORTHO08HWII, ORTHO053E, and ORTHO066E exhibited higher Mash distances (0.085–0.094), signifying greater sequence divergence. This suggests the coexistence of a dominant, conserved IncFIB(K) lineage with more divergent plasmid variants, reflecting both clonal spread and underlying genomic plasticity.
Col440I plasmids showed a combination of conserved and slightly divergent sequences. An exact match (Mash distance = 0) between isolates ID014 and ID064 suggests recent common ancestry or clonal dissemination. Other pairwise distances ranged from 0.024 to 0.035, indicating limited sequence variation potentially driven by recombination or gene acquisition. These findings support a largely conserved Col440I plasmid population with evidence of close epidemiological links among clinical isolates.