
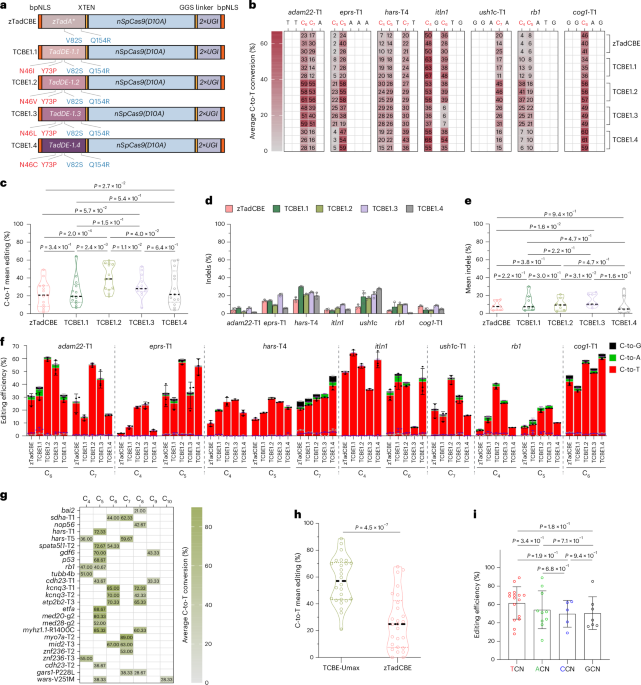
Development of highly efficient TadA-derived CBE editors
Our goal was to develop high-efficiency next-generation TadA-derived CBE editors to enable biallelic editing in vivo. Recently, phage-assisted evolution was employed to develop CBE6s from a TadA-mediated dual cytosine and adenine base editor14. Through this process, mutations at positions N46 and Y73 were identified in TadA, which not only eliminated residual A•T-to-G•C editing but also enhanced C•G-to-T•A editing while broadening sequence-context compatibility14. Our previous research further demonstrated that the V82S and Q154R mutations in TadA enhance the C-to-T editing efficiency of zTadCBE in zebrafish13. However, whether these mutations are compatible with N46 and Y73, and whether their combination could further enhance CBE activity, remain unknown.
To address this question, we constructed four novel editors incorporating different combinations of these mutations. To optimize functionality in zebrafish, all editors underwent codon optimization and were designated as follows: TCBE-1.1 (N46I, Y73P, V82S and Q154R), TCBE-1.2 (N46V, Y73P, V82S and Q154R), TCBE-1.3 (N46L, Y73P, V82S and Q154R) and TCBE-1.4 (N46C, Y73P, V82S and Q154C); all four editors were constructed on the basis of the TadA-derived dual editor (TadDE), similar to what was done in the previous study14 (Fig. 1a). To evaluate the performance of these newly designed enzymes, we selected seven target sites with varying sequence contexts and assessed their C-to-T editing efficiencies using high-throughput sequencing. All editors exhibited editing activity at the target cytosines, albeit with variable efficiencies (Fig. 1b). Among these, TCBE-1.2 demonstrated the highest editing efficiency, followed by TCBE-1.4, whereas TCBE-1.1 and TCBE-1.3 showed no significant improvements compared to the original base editor, zTadCBE (Fig. 1c). Regarding indel formation, apart from TCBE-1.3, the remaining three editors did not exhibit a notable increase compared to zTadCBE (Fig. 1d,e). Low-frequency undesired by-products (C-to-A and C-to-G conversions) were detected at a frequency of less than 6% (Fig. 1f), demonstrating high editing specificity for all editors.
Fig. 1: Screening and characterization of hyper TadA-derived cytosine base editor TCBE-Umax.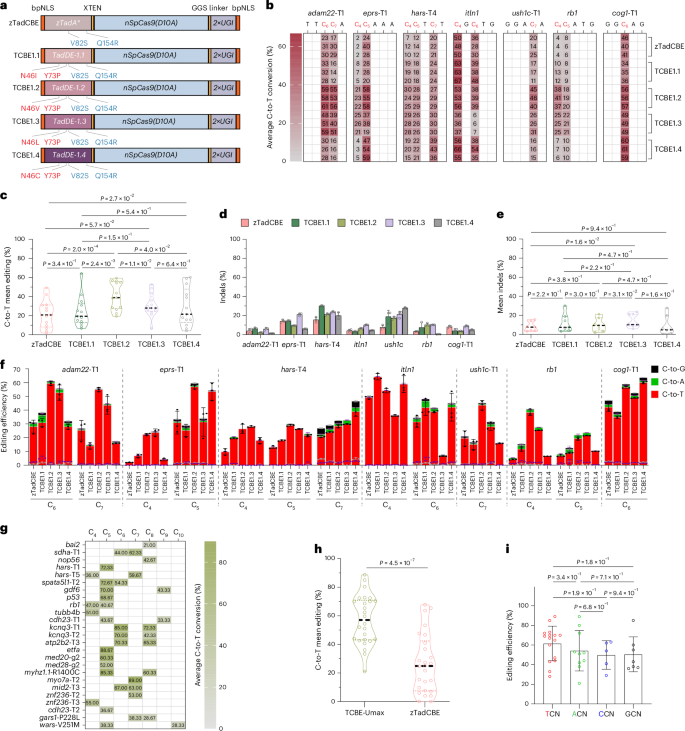
a, Schematic representation of constructs for five TadA-derived CBEs. Components include: bpNLS (bipartite nuclear localization signal), various deaminase mutations (zTadA*, TadA*-1.1 through TadA*-1.4), XTEN (32-amino-acid flexible linker), nSpCas9 (SpCas9 D10A nickase), GGS linker (GGSSGGS amino acid sequence) and UGI (uracil glycosylase inhibitor). b, Heat maps depicting C-to-T conversion frequencies at each position for indicated sites across five editors: zTadCBE, TCBE-1.1, TCBE-1.2, TCBE-1.3 and TCBE-1.4. Data represent the mean of three independent biological replicates. c, Mean editing efficiency comparison of zTadCBE, TCBE-1.1, TCBE-1.2, TCBE-1.3 and TCBE-1.4 editors based on data from b. Individual data points show mean editing activity per site, with the central dashed line indicating overall mean efficiency. Statistical significance was determined using a nonparametric two-sided Wilcoxon matched-pairs signed-rank test (P values shown above the violin plot). d, Indel frequencies of the indicated sites for zTadCBE, TCBE-1.1, TCBE-1.2, TCBE-1.3 and TCBE-1.4. Data are presented as mean ± s.d., calculated from three biological replicates. e, Comprehensive indel frequency analysis for zTadCBE, TCBE-1.1, TCBE-1.2, TCBE-1.3 and TCBE-1.4. Each data point represents the mean indel frequency per site, with the central dashed line indicating the overall mean. Statistical significance was assessed using the nonparametric two-sided Wilcoxon matched-pairs signed-rank test (P values indicated above the violin plot). f, Product purity analysis at 7 endogenous genomic loci. Values represent mean ± s.d. of three independent experiments. g, Editing efficiency of TCBE-Umax at the additional 25 endogenous targets across 17 genes. Data represent the mean of three independent biological replicates. h, Assessment of the mean editing efficiency of zTadCBE and TCBE-Umax using 17 gRNAs targeting NGG PAMs. Each data point represents the average editing activity at a specific site. The central dashed line represents the mean of all data points. P values are displayed at the top of the violin plot. Statistical analysis was performed using a two-tailed paired t-test (P values indicated). i, Base editing efficiency of TCBE-Umax systems at target C in various NCN sequence contexts. Each data point represents the mean editing activity of an individual genomic site. Error bars represent the mean ± s.d. calculated across all tested sites within each group. Statistical analysis was performed using a two-tailed paired t-test (P values indicated).
Given the superior activity displayed by TCBE-1.2, we named it TCBE-Umax and focused on its further characterization and optimization. To ensure that the optimized editor exhibits broad applicability across diverse genomic loci, we further characterized the editing profile of TCBE-Umax at an additional 25 endogenous zebrafish target sites with NGG PAM, covering 17 genes of interest. All tested sites exhibited high editing efficiencies (up to 89%) within the primary editing window (positions 4–8 of the protospacer), resulting in the expected C-to-T conversions (Fig.1g). Compared to the previous tool, zTadCBE, TCBE-Umax demonstrated a more than double (~2-fold) improvement in average editing efficiency (Fig. 1h and Supplementary Fig. 1). Because traditional CBEs have strong TC sequence-context bias, we also assessed the sequence-context preference of TCBE-Umax, which exhibited consistent C•G-to-T•A editing efficiency across all tested sequence contexts (Fig. 1i).
To further characterize the base specificity of TCBE-Umax in vivo, we asked whether this editor retains any residual A-to-G editing activity inherited from the TadA-derived deaminase. Our previous studies reported that zTadCBE exhibited minimal A-to-G activity (<10%) at certain sites in zebrafish13. While these unintended edits can be diluted over successive generations, these edits pose analytical challenges, particularly when using the F0 generation for direct phenotype assessment. We therefore compared TCBE-Umax with zTadCBE at three loci (bai2, hars-T1 and nop56) where zTadCBE mediates low A-to-G conversions. As expected, zTadCBE induced clear A-to-G substitutions at the target adenines, whereas TCBE-Umax did not generate detectable A-to-G edits above background at any of the tested sites (Supplementary Fig.2).
High editing activity can increase off-target effects; to assess this, we first examined the guide RNA (gRNA)-dependent off-target activity of TCBE-Umax at the top three predicted sites for the p53, kcnq3-T2 and spata5l1-T2 gRNAs. High-throughput sequencing revealed that TCBE-Umax showed no detectable editing at the kcnq3-T2 and spata5l1-T2 loci, while exhibiting slightly reduced off-target effects (<10%) for the p53-targeting gRNA (Supplementary Fig. 3a). Next, we investigated whether TCBE-Umax induces gRNA-independent off-target modifications across the genome. To do this, we co-injected TCBE-Umax with its respective gRNA, along with a catalytically inactive Staphylococcus aureus-Cas9 system (dSaCas9) and gRNAs targeting R-loop sites 1–3. The dSaCas9 system facilitated the formation of R-loops, short single-stranded DNA segments that could potentially serve as substrates for TCBE-Umax (Supplementary Fig. 3b). Next-generation sequencing analysis showed that TCBE-Umax maintained comparable on-target editing efficiency regardless of dSaCas9 presence (Supplementary Fig. 3c). Regarding off-target activity, TCBE-Umax exhibited negligible editing at all three R-loop sites compared to the control group (Supplementary Fig. 3d), highlighting its high specificity.
Expanding the targeting range of TCBE-Umax
Having shown that our TCBE-Umax editors are substantially more efficient than previous TCBEs, we next set out to further improve editing versatility by expanding their targeting scope. Base editors derived from Streptococcus pyogenes Cas9 (SpCas9) are constrained by both the requirement of the PAM—a short 5′-NGG-3′ DNA sequence adjacent to the cleavage site—and their restricted base editing to a narrow window of 4–8 bases distal to the PAM. Engineered SpCas9 enzymes, such as SpRY and SpG, have been developed to exhibit more flexible PAM recognition and are compatible with single-base editing systems in zebrafish3,15,16. These modified enzymes enable the recognition of atypical 5′-NNN-3′ PAM sequences, broadening the targetable genomic range of base editors.
To expand the targeting scope of TCBE-Umax-mediated cytosine base editing, we replaced the nSpCas9 in the TCBE-Umax construct with nSpRYCas9, generating TCBE-Umax-SpRY (Fig. 2a). To evaluate its C-to-T editing efficiency at NNN PAM sites in zebrafish, we selected 18 loci with atypical PAM sequences and co-injected single guide RNAs (sgRNAs) with TCBE-Umax-SpRY-encoding mRNA into one-cell-stage embryos. Sequencing results revealed C-to-T conversions at all 18 loci, albeit with varying efficiencies (Fig. 2b). Compared to our previously developed zTadCBE-SpRY13, TCBE-Umax-SpRY demonstrated an average 1.7-fold increase in C-to-T editing efficiency (Fig. 2c,d). These findings confirm that TadA-based C-to-T base editing is compatible with nSpRYCas9, enabling efficient editing across a broad range of PAM sequences in zebrafish and significantly expanding the targeting scope of the base editor.
Fig. 2: Expanding the targeting range of different TCBE-Umax editors.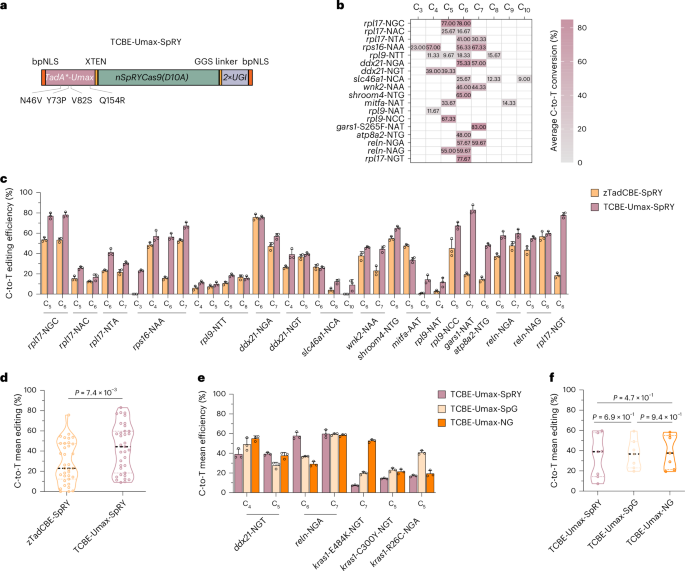
a, Schematics of the TCBE-Umax-SpRY editor used for cytosine base editing in zebrafish. b, Summary of TCBE-Umax-SpRY editing efficiency across 18 NNN PAM sites in 11 genes. Heat maps depict C-to-T conversion frequencies at each position for the indicated sites. Data represent the mean of three independent biological replicates. c, Comparison of editing efficiencies between zTadCBE-SpRY and TCBE-Umax-SpRY using 18 gRNAs targeting NNN PAMs. Numbers indicate the position of the edited base within each gRNA. Data represent mean ± s.d. of three biological replicates. d, Analysis of mean editing efficiency comparing zTadCBE-SpRY and TCBE-Umax-SpRY based on data from c. Individual data points show mean editing efficiency per site, with the overall mean indicated by a central dashed line. P values from two-tailed paired t-tests are shown above the violin plot. e, Comparison of editing efficiencies among TCBE-Umax-SpRY, TCBE-Umax-SpG and TCBE-Umax-NG at five different loci with NGN PAM. Numbers indicate the editing base position within the gRNA. Values represent mean ± s.d. (n = 3 biological replicates). f, Summary of editing efficiency for TCBE-Umax-SpRY, TCBE-Umax-SpG and TCBE-Umax-NG across five tested different loci with NGN PAM based on data from e. Each data point represents the mean editing efficiency of an individual locus, while the central dashed line indicates the overall mean. A nonparametric two-sided Wilcoxon matched-pairs signed-rank test was performed (P values indicated above the violin plot).
Whereas several Cas9 enzymes, including SpGCas9, Cas9-NG and ScCas9, have been reported to function in zebrafish, only the SpRY enzyme has been demonstrated to be compatible with CBEs in this model organism15,17,18. Similar to a previous report18, our experiments showed that CBE4-SpG exhibited extremely low activity in zebrafish, rendering it unsuitable for practical applications in zebrafish. To further enhance the diversity of base editing tools and expand options for editing specific loci, we constructed two additional editors, TCBE-Umax-SpG and TCBE-Umax-NG, both of which require only a single G in the PAM. Upon evaluating five such NGN PAM sites, these two editors exhibited variable activities across different loci, confirming their compatibility with the TCBE-Umax system (Fig. 2e). While their average activity was comparable to that of TCBE-Umax-SpRY (Fig. 2f), this editor demonstrated superior editing efficiency at specific loci. For instance, at the kras E484K-NGT locus, TCBE-Umax-NG achieved an editing efficiency of 53%, whereas TCBE-Umax-SpRY exhibited only 7% efficiency. Similarly, at the kras R26C-NGA locus, TCBE-Umax-SpG outperformed the other two editors (Fig. 2e). These results highlight that, in non-NGG PAM contexts, selecting different Cas9 editors can effectively maximize base editing efficiency at specific loci. We further demonstrated that all the TCBE-Umax editors discussed above achieved high germline targeting efficiency and transmission rates (Supplementary Table 1), highlighting their strong capability for precise and efficient base editing.
Characterization of two TCBE-Umax editors with reduced indel generation and expanded targeting range
While evaluating TCBE-Umax activity, we observed that approximately one-third (8 out of 25) of the target regions exhibited overlapping peaks near the target sites in the Sanger sequencing chromatograms, suggesting the presence of indels alongside cytosine base editing (Supplementary Fig. 4). The relatively high frequency of indels compromises the precision of this tool, particularly when assessing variant pathogenicity in the F0 generation. Therefore, we set out to develop an editor that minimizes indel formation while maintaining high editing efficiency. CRISPR–Cas9 naturally contains multiple sites in its protein sequence where inteins can be inserted without disrupting function19. A previous study reported that the amino acid at position 1,054 within the RuvC domain of Cas9 is structurally positioned on the protein surface, where it exhibits conformational flexibility and lies in proximity to the non-target DNA strand, making it potentially susceptible to deamination20. Given this, we hypothesized that altering the relative position of the deaminase and Cas9 might modify the intrinsic properties of the base editor. To test this hypothesis, we inserted the deaminase at position 1,054 within Cas9, without linkers, directly fusing it to Cas9 to create a new editor, which we designated as TCBE-Umax-ex1 (Fig. 3a,b). Initial Sanger sequencing at two sites with high indel frequencies following TCBE-Umax editing led to an unexpected discovery: TCBE-Umax-ex1 significantly reduced indel formation (Supplementary Fig. 5). To systematically and quantitatively evaluate its effect on indel generation, we selected 8 loci with high indel frequencies by TCBE-Umax editing and performed high-throughput sequencing. The results demonstrated a substantial reduction in indel generation across all tested sites, with indel levels decreasing to approximately one-fourth of those observed with TCBE-Umax (Supplementary Fig. 6). In addition, analysis of by-products revealed that TCBE-Umax-ex1 also maintained a low level of undesired base conversions (Supplementary Fig. 7). Notably, on-target editing activity remained comparable to or slightly improved (Fig. 3c) over that of TCBE-Umax. Meanwhile, TCBE-Umax-ex1 exhibited a slight shift in its editing window compared to TCBE-Umax at these 8 loci. We, therefore, assessed its activity at an additional 13 target sites (Fig. 3d). The final analysis revealed that TCBE-Umax-ex1 has an editing window spanning positions 3 to 12, with peak activity concentrated between positions 4 and 10, whereas TCBE-Umax primarily edits positions 4–8 (Fig. 3e).
Fig. 3: Characterization of TCBE-Umax editors with expanded targeting range and reduced indel formation.
a, Schematic representation of the TCBE-Umax-ex1 construct. b, AlphaFold2-predicted structure of TCBE-Umax-ex1. c, Comparative editing efficiency of TCBE-Umax and TCBE-Umax-ex1 at eight endogenous genomic sites in zebrafish. The heat map shows average editing percentages of three independent biological replicates. d, TCBE-Umax-ex1 editing efficiency across 13 endogenous targets in 13 distinct genes. Heat map depicts C-to-T conversion frequencies at each position for indicated sites. Data represent the mean of three independent biological replicates. e, Efficiency and targeting window comparison between TCBE-Umax and TCBE-Umax-ex1. Data points represent average editing activity per site. The targeting windows, measured from the 5′ to the 3′ termini of the targeting sites, are highlighted in green for TCBE-Umax (positions 4–10) and in blue for TCBE-Umax-ex1 (positions 3–12). Analysis is based on data from three independent experiments. f, Schematic representation of the TCBE-Umax-ex2 construct, designed to modify the cytosine base editing window. g, AlphaFold2-predicted structure of TCBE-Umax-ex2. h, TCBE-Umax-ex2 editing efficiency across 15 endogenous targets in 12 genes. The heat map displays average editing percentages of three independent biological replicates. i, Efficiency and targeting window comparison between TCBE-Umax and TCBE-Umax-ex2. Data points represent average editing activity per site. The targeting windows, measured from the 5′ to the 3′ terminal of the targeting site, are highlighted in green for TCBE-Umax (positions 4–10) and red for TCBE-Umax-ex2 (positions 4–16). Analysis is based on data from three independent experiments.
While TCBE-Umax-ex1 expands the editing window, its activity remains distant from the PAM site. To overcome this limitation, we developed a second editor, TCBE-Umax-ex2 (Fig. 3f,g). In TCBE-Umax-ex2, the HNH domain of Cas9 was deleted. A GGS linker was introduced to connect SpCas9 at position S793 to the N terminus of TCBE-Umax, while an SGG linker linked the C terminus of TCBE-Umax to SpCas9 at position R919. To assess its editing window, we evaluated TCBE-Umax-ex2 across 15 target sites in zebrafish. (Fig. 3h). Compared to conventional TCBE-Umax, TCBE-Umax-ex2 shifted the editing window towards the PAM-proximal region of the protospacer, extending from positions 5 to 16, with peak activity at positions 13–15 (Fig. 3i). By-product analysis using high-throughput sequencing revealed that this tool is highly specific, displaying minimal levels of undesired base conversions (Supplementary Fig. 8). Further indel analysis demonstrated that TCBE-Umax-ex2 also induces very low levels of indels (Supplementary Fig. 9). Notably, TCBE-Umax-ex2 exhibited enhanced editing efficiency at specific loci compared to zTadCBE-ex2 from our previous study13 (Supplementary Fig. 10), further expanding its utility for precise base editing applications. Our findings demonstrate that these two TCBE-Umax editors effectively address key limitations in targeting range and reduce unwanted indel formation. Their optimized designs provide greater flexibility in selecting efficient editors for specific loci, offering more precise tools for pathogenicity assessments and broader applications in base editing, particularly at sites prone to high indel rates.
Characterization of three TCBE-Umax editors with enhanced precision
Expanding the editing window of base editors enhances their utility in studying SNVs. However, broader editing windows also increase the risk of bystander edits, introducing confounding factors in the assessment of SNV pathogenicity. Therefore, developing base editors with narrower editing windows while maintaining high activity remains a critical challenge, particularly for clinical applications and functional testing. Several strategies have been used to restrict the editing window of base editors. These include structure-guided engineering to modify deaminase properties, optimizing the deaminase–Cas9 linkage to enhance editing precision, and using library-assisted protein evolution to generate editors with enhanced specificity21,22,23.
To assess whether specific modifications could effectively limit the editing window of TCBE-Umax in zebrafish, we developed three editors: TCBE-Umax-rest1, featuring an N108Q mutation in the deaminase domain; TCBE-Umax-rest2, with a linker deletion between the deaminase and Cas9 domains; and TCBE-Umax-rest3, incorporating three targeted mutations (N119D, N122H and G125D) known to enhance specificity. These changes were chosen on the basis of their ability to narrow editing windows in cell culture systems, enabling us to evaluate their impact in a vertebrate in vivo model (Fig. 4a). High-throughput sequencing at 8 target sites revealed that the 3 editors effectively narrowed the editing window to varying degrees (Fig. 4b). Among them, TCBE-Umax-rest2 demonstrated the most favourable editing profile, with activity centred at C4–C7, and the lowest bystander editing efficiency at 6 of the 8 loci. TCBE-Umax-rest1 ranked second in performance, whereas TCBE-Umax-rest3 did not exhibit a significant advantage over the other two editors (Fig. 4b). Regarding on-target efficiency, despite reduced activity, TCBE-Umax-rest2 retains ~75% of TCBE-Umax activity. TCBE-Umax-rest3 ranked second in activity. TCBE-Umax-rest1 exhibited a significant reduction in activity but retained a substantial proportion (44%) of the efficiency of the original tool (Fig. 4c). In terms of indel formation, only TCBE-Umax-rest2 showed a significant reduction, while the other two editors exhibited no notable difference from TCBE-Umax (Supplementary Fig. 11). Notably, TCBE-Umax-rest1 does not mirror the behaviour of ABE-Umax-rest1, where the same N108 mutation improves precision but simultaneously elevates indel formation (Supplementary Fig. 12). In our TCBE-Umax framework, TCBE-Umax-rest1 achieves a more restricted editing window while maintaining indel levels that remain acceptable, highlighting that identical mutations can have distinct functional consequences in different base editor architectures and therefore require empirical evaluation in each context. By-product analysis revealed that TCBE-Umax-rest1, -rest2 and -rest3 each displayed noticeable C-to-G conversion activity at specific sites (hars-T4-C7, atp2b2-T3-C6 and kcnq3-T1-C6) containing the TCT motif (Supplementary Fig. 13). This observation is consistent with findings reported in previous studies24. Analysis of editing window profiles indicated that TCBE-Umax-rest1 primarily targeted C5–C7 bases, whereas TCBE-Umax-rest2 exhibited a broader editing range spanning C4–C7 bases. As for TCBE-Umax-rest3, minimal activity was consistently detected at positions C8–C9 at 6 of the 8 loci (Fig. 4d). Based on these findings, TCBE-Umax-rest2 emerges as the preferred, best-performing editor for precision editing applications, provided that C4 editing does not interfere with experimental outcomes. If C4 editing is undesirable, TCBE-Umax-rest1 presents a viable alternative. These results underscore the potential of structure-guided engineering and linker optimization in refining CBE editing specificity, offering enhanced precision for single-base editing applications in zebrafish and other model systems.
Fig. 4: Characteristics of TCBE-Umax editors as precise genome editing tools in zebrafish.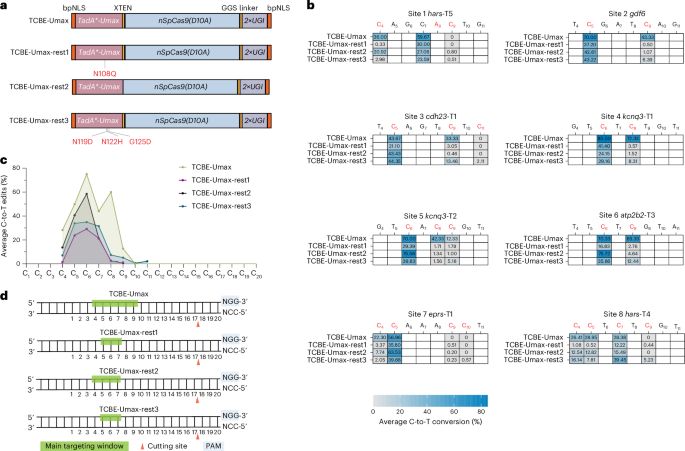
a, Schematic representation of TCBE-Umax editors (TCBE-Umax, TCBE-Umax-rest1, TCBE-UMax-rest2 and TCBE-UMax-rest3) designed for precise cytosine base editing. TCBE-Umax-rest1 incorporates an N108Q mutation in the deaminase domain; for TCBE-Umax-rest2, the XTEN linker between the deaminase and Cas9 domains was deleted; TCBE-Umax-rest3 harbours three rationally designed mutations (N119D, N122H and G125D) within the deaminase. b, Editing efficiency comparison of TCBE-Umax, TCBE-Umax-rest1, TCBE-Umax-rest2 and TCBE-Umax-rest3 at eight endogenous genomic sites in zebrafish. The heat map displays average editing percentages of three independent biological replicates. c, Comparative analysis of average C-to-T editing efficiency among TCBE-Umax, TCBE-Umax-rest1, TCBE-Umax-rest2 and TCBE-Umax-rest3 editors at the eight target sites shown in b. d, Schematic illustration of the editing window for TCBE-Umax tools. Green highlighting indicates the main editing windows. The SpCas9 cutting site is marked by a red triangle, with the PAM sequence and its complement highlighted in light blue.
Functional analysis of candidate disease genes in the F0 generation using TCBE-Umax
CRISPR–Cas9-mediated genome editing enables the direct creation of homozygous knockout organisms without requiring multiple generations of breeding through its capacity to generate biallelic mutations25,26,27. This capability is particularly advantageous in zebrafish research, where phenotypic analysis can be conducted directly in the F0 generation with unprecedented speed and efficiency25,26,27. We aimed to investigate whether biallelic mutations in disease-causing genes can be generated using our TCBE editor and whether the resulting phenotypes can be screened in the F0 generation. To test this approach, we selected zebrafish orthologues of six human disease genes (myo7aa, hars, med4, nup160, etfa and myhz1.1), which, when mutated, cause diseases affecting multiple tissues. MYO7A, a motor protein essential for inner ear and retinal function, causes Usher syndrome type 1B and non-syndromic hearing loss when mutated28. We induced a premature stop codon in zebrafish myo7aa by changing C→T (CGA (Arg) to TGA (stop)) base using TCBE-Umax, achieving 100% base conversion. We analysed lateral line hair cell function utilizing YO-PRO-1 dye, which is taken up by mechanotransduction channels. Control animals showed normal hair cell development, while edited animals showed compromised hair cell function, demonstrated by the absence of YO-PRO-1 dye (Fig. 5a). Similarly, we generated premature stop codons using the TCBE-Umax editor in the zebrafish orthologues of HARS1, MED4 and NUP160. HARS1 encodes a transfer RNA synthetase crucial for protein synthesis associated with Usher syndrome type 3B and Charcot-Marie-Tooth disease type 2W29. MED4, a transcriptional regulator in the mediator complex, is associated with developmental delays and congenital anomalies30. NUP160, a nuclear pore protein involved in molecular transport, is associated with neurodevelopmental disorders and steroid-resistant nephrotic syndrome31. Mutations in these three genes showed early developmental abnormalities (Fig. 5b–d). In addition, we introduced a premature stop codon in etfa, a mitochondrial protein essential for fatty acid metabolism, which causes multiple Acyl-CoA dehydrogenase deficiency32. Control and edited larvae were analysed using whole-mount staining with oil red O (ORO) to assess lipid accumulation in the liver. Indeed, edited animals showed increased lipid accumulation in the liver, suggestive of steatosis and hyperlipidemia (Fig. 5e). Finally, we utilized TCBE-Umax to create a missense mutation in the myhz1.1 gene by converting C→T, which changes Arg (CGT) to Cys (TGT). The zebrafish gene myhz1.1 encodes a protein predicted to be part of the myosin II complex, and its human orthologues have been implicated in multiple muscle tissue diseases33. TCBE-Umax induced the R1398C mutation with high efficiency. Indeed, edited zebrafish larvae (R1398C) showed abnormal muscle development by 5 days post fertilization (dpf) compared to control animals, demonstrated by fluorescent-conjugated phalloidin staining (Fig. 5f). In conclusion, we were able to generate both loss-of-function and missense mutation in the orthologues of various human disease genes and analysed the phenotypes in injected animals (F0), thereby facilitating rapid functional analysis of candidate disease genes.
Fig. 5: Functional evaluation of candidate disease genes in the F0 generation using TCBE-Umax.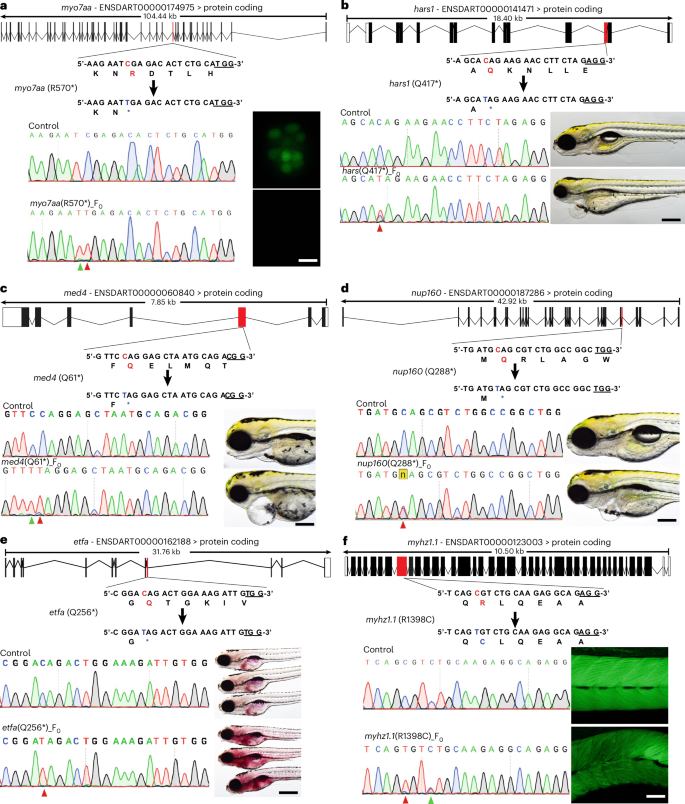
a, Schematic representation of the myo7aa (R570*) target locus. The target sequence is shown with the PAM site underlined. Red highlighting indicates the original nucleotide and amino acid positions, while blue highlighting shows the expected nucleotide and amino acid substitutions. The figure presents sequencing results and phenotypes of myo7aa (R570*) mutations induced by TCBE-Umax. In the Sanger sequencing chromatograms, red arrowheads indicate the expected nucleotide substitutions, while green arrowheads mark bystander base substitutions. Adjacent to the chromatograms are images of neuromast hair cells labelled with YO-PRO-1 live dye (green). The images are representative of three independent experiments. Scale bars, 10 µm. b, Schematic representation of the hars (Q417*) target site. The target sequence is shown with an underlined PAM region. Red highlights the original nucleotide and amino acid, while blue highlights the anticipated nucleotide and amino acid substitutions. The sequencing data and phenotypic observations from TCBE-Umax-mediated editing of hars (Q417*) are presented. A red arrowhead indicates the expected nucleotide changes. Animals edited using TCBE-Umax exhibited developmental defects during the early stages. The images are representative of three independent experiments. Scale bar, 200 μm. c, A schematic diagram illustrating the med4 (Q61*) target site. The diagram shows the target sequence with its PAM sequence underlined. Red highlighting marks the original nucleotide and amino acid, while blue indicates their intended modifications. The results include Sanger sequencing chromatograms and phenotype observations from TCBE-Umax editing. Red arrowheads indicate the expected nucleotide changes, and green arrowheads mark unintended bystander substitutions in the chromatograms. Animals edited using TCBE-Umax exhibited developmental abnormalities during the early stages. The images are representative of three independent experiments. Scale bar, 200 μm. d, Schematic representation of the nup160 (Q288*) gene locus. The target sequence is displayed with the PAM underlined. The original nucleotide and amino acid (red) and desired changes (blue) are highlighted. Sequencing results and phenotypic effects of the TCBE-Umax-induced nup160 (Q288*) mutation are presented. A red arrowhead marks expected nucleotide substitutions. Edited animals exhibiting early-stage developmental defects were consistently observed. The images are representative of three independent experiments. Scale bar, 200 μm. e, Schematic diagram of the etfa (Q256*) target locus. The target sequence is shown with the PAM underlined. The original nucleotide and amino acid (red) and expected changes (blue) are highlighted. Sequencing results and phenotypic effects of etfa (Q256*) mutations induced by TCBE-Umax are presented. A red arrowhead marks expected nucleotide substitutions. Edited animals exhibited increased lipid accumulation, a consistent finding across three experiments. Scale bar, 200 μm. f, Schematic diagram of the myhz1.1 (R1398C) target locus. The target sequence is displayed with the PAM underlined. Original (red) and expected (blue) nucleotide and amino acid changes are highlighted. Sequencing results and phenotypic observations of myhz1.1 (R1398C) mutations induced by TCBE-Umax are shown. The red arrowhead marks expected nucleotide substitutions, while a green arrowhead in Sanger chromatograms indicates bystander substitutions. Edited animals exhibited abnormal muscular development, as observed through fluorescent-conjugated phalloidin staining. The images are representative of three independent experiments. Scale bar, 100 μm.
Rapid functional validation of genetic variants associated with genetic disorders
The completion of genome sequencing projects has revealed countless single-nucleotide polymorphisms (SNPs), predominantly missense mutations. Given the high efficiency and specificity of TCBE-Umax editors, we investigated their potential for functional analysis of these genetic variants, particularly ‘variants of uncertain significance’ (VUS), which are not known to be either benign or disease causing.
Single-base editing technologies (CBE or ABE) enable precise single-nucleotide modifications; however, their use in organism-wide studies is limited by variable editing efficiency across different genetic locations, sequence-context dependencies and inherent technical constraints. We set out to utilize the TCBE-Umax system, which boasts high efficiency and minimal sequence-context bias, to assess the pathogenicity of VUS in Myo7a and Cdh23, two key proteins implicated in hereditary hearing loss. Cadherin-23, encoded by the CDH23 gene, is essential for forming stereocilia tip links in sensory hair cells. Mutations in CDH23 can cause either Usher syndrome type 1D or non-syndromic deafness (DFNB12)34, both of which impair mechanotransduction and hearing. Similarly, the MYO7A gene encodes myosin VIIA, an actin-based motor protein crucial for the organization of stereocilia, intracellular trafficking and mechanotransduction in hair cells. Mutations in MYO7A result in Usher syndrome type 1B or DFNB2, causing progressive hearing loss and vestibular dysfunction35. In zebrafish, loss-of-function mutations in either cdh23 or myo7aa disrupt hair cell function and stereocilia integrity, leading to impaired mechanosensory signalling and loss of the acoustic startle response (AEBR)36,37. For these studies, we utilized the zebrafish lateral line system, which enables rapid assessment of hair cell function in an intact organism. The molecular machinery of hair cells is highly conserved between zebrafish and humans, making findings from this model system directly relevant to human hearing loss research. As described earlier, we employed the YO-PRO-1 live dye uptake assay to quantitatively measure mechanotransduction function, while the accessibility of hair cells allows direct observation of phenotypic changes (Fig. 6a).
Fig. 6: Rapid functional evaluation of VUS associated with hearing loss.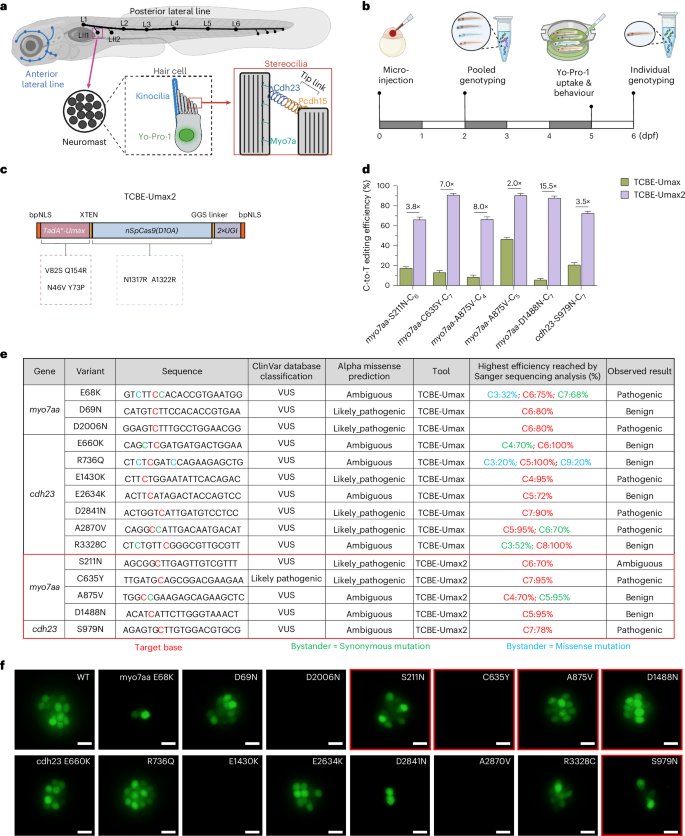
a, Schematic representation of the zebrafish lateral line sensory system which consists of clusters of mechanosensory hair cells known as neuromasts. These lateral line hair cells are functionally and molecularly similar to those in the zebrafish inner ear, making them an excellent model for studying hearing function. Functional hair cells in live zebrafish embryos can be labelled using YO-PRO-1 dye, taken up through hair cell mechanotransduction channels. A schematic of the stereocilia structure highlights key molecular components, including Cdh23 and Myo7a. b, Experimental workflow for the rapid assessment of VUS pathogenicity using zebrafish. c, Schematic diagrams of TCBE-Umax2 constructs, designed to enhance cytosine base editing efficiency. d, Comparison of editing efficiencies between TCBE-Umax and TCBE-Umax2 at five target sites. Fold changes are indicated above each group. Data are presented as mean ± s.d. of three biological replicates. e, Summary of all 15 VUS target sites, including their editing efficiencies and observed outcomes. f, Yo-PRO-1 staining results corresponding to the 15 VUS target sites. Three independent experiments yielded similar results. Scale bars, 10 µm. Sites edited by TCBE-Umax2 are highlighted in red boxes. Panels a and b were created in BioRender. Varshney, G. (2025) https://BioRender.com/7hrho92.
We selected 15 missense mutations (C→T) in the MYO7A and CDH23 genes from the ClinVar database for functional evaluation (Supplementary Figs. 14–16). Of these mutations, 14 were SNPs classified as VUS, while 1 was considered likely pathogenic and served as our positive control. Our screening pipeline began with co-injecting TCBE-Umax mRNA and target variant-specific gRNAs into zebrafish embryos at the single-cell stage. After 2 dpf, we evaluated editing efficiency by analysing pooled genomic DNA from 10 random embryos using PCR and Sanger sequencing, with variants showing greater than 50% efficiency advancing to further testing. At 5 dpf, we assessed morphology and performed acoustic startle response tests, followed by YO-PRO-1 live staining at 6 dpf to evaluate hair cell function. Finally, we conducted single-embryo genotyping for variants exhibiting phenotypic effects on groups of 5–12 embryos to validate editing outcomes and determine the correlation between editing efficiency and phenotype severity. Specifically, variants showing positive phenotypes in both assays underwent genotyping with 5 embryos per group, while cases with phenotypic variability required genotyping of 12 embryos to accurately determine variant pathogenicity through the correlation of editing efficiency with phenotypic severity (Fig. 6b).
Initial assessment of editing efficiency in pooled embryos at 2 dpf revealed that 10 of the 15 sites achieved average editing efficiency above 50% (Supplementary Table 2). Among these 10 variants, we performed acoustic response assays, and 5 of the variants did not respond to sound stimuli, strongly indicating pathogenicity, a finding subsequently confirmed through YO-PRO-1 staining and genotyping (Fig. 6e,f, and Supplementary Fig. 17 and Table 2). The remaining 5 high-efficiency variants underwent YO-PRO-1 staining and genotyping analysis in 12 individual embryos per group. Despite high editing efficiency (>50%, reaching 100% at some sites), these embryos exhibited staining patterns comparable to those of the controls, suggesting that these variants are probably benign (Fig. 6e,f, and Supplementary Fig. 17 and Table 2).
As mentioned above, in our initial screening, 5 variants displayed low editing efficiency (<50%), complicating accurate pathogenicity assessment. To address this limitation, we further optimized TCBE-Umax to improve its efficiency for F0-generation zebrafish studies. Recent studies exploring nCas9 engineering to enhance base editing have revealed that N1317R + A1322R mutations in Cas9 improve target DNA binding, showing enhanced activity at 5 out of 6 tested sites38. On the basis of these findings, we incorporated these mutations into the TCBE-Umax editor to create the TCBE-Umax2 editor (Fig. 6c).
TCBE-Umax2 significantly improved on-target editing efficiency across all five previously low-efficiency sites, achieving 2- to 15.5-fold increases compared to TCBE-Umax and raising all average editing efficiencies above 50% (Fig. 6d). Subsequent phenotypic and genotypic assessment of these variants revealed another two as pathogenic, exhibiting uniform loss of acoustic response and reduced YO-PRO-1-positive hair cells, while two others showed normal function, confirming them as benign (Fig. 6e,f, and Supplementary Fig.17 and Table 2). One variant, myo7aa-S211N, presented an intriguing case: despite consistently high editing efficiency (>50%) across all 12 tested embryos, YO-PRO-1 staining results varied markedly as few embryos showed substantial signal reduction, while others remained normal (Supplementary Table 2). This variability suggests the need for further evaluation in stable germline-transmitted zebrafish lines, highlighting that single mutations can produce varying phenotypic manifestations, as observed in human patients.
It is worth noting that TCBE-Umax2 is not a universal solution. When benchmarked across a broader panel of endogenous zebrafish loci, TCBE-Umax2 displayed more variable C-to-T editing efficiencies than TCBE-Umax and was often associated with elevated indel frequencies as well as detectable gRNA-dependent off-target edits at some predicted sites (Supplementary Figs.18–20). Nonetheless, the modular design of TCBE-Umax2, which combines a hyperactive deaminase with an activity-enhanced Cas9, allowed us to reuse the same three precision-enhancing mutations previously engineered into TCBE-Umax. The resulting variants maintained efficient C-to-T editing while restricting activity to narrow windows across representative targets (Supplementary Fig. 21).
Taken together, our optimized strategy, using TCBE-Umax as a first-line editor and TCBE-Umax2-based variants as complementary tools for refractory loci, enabled rapid functional assessment of 14 VUS in zebrafish in the F0 generation, establishing a general framework for large-scale, organismal-level evaluation of variant pathogenicity.
Strategy for selecting the appropriate TadA-based CBE
We have developed several TCBE-Umax editors, each with unique characteristics (Supplementary Fig. 22), and selecting the most suitable TadA-based CBE is crucial for achieving optimal results. Our selection strategy, outlined in Fig. 7, begins by checking for a GG motif 6–21 base pairs downstream of the target cytosine to meet the NGG PAM requirement. If present, we prioritize tools recognizing NGG PAM sequences, favouring TCBE-Umax (editing window: 4–10 bases on the protospacer), TCBE-Umax-ex1 (window: 3–12 bases), or TCBE-Umax-ex2 (window: 4–16 bases) for maximum efficiency, with TCBE-Umax-ex1 being the top choice due to its minimal or absent indel induction. If TCBE-Umax shows suboptimal activity at a given site, TCBE-Umax2 may be considered as an alternative to improve editing efficiency. For applications prioritizing precision, TCBE-Umax-rest1 (window: 5–7) or TCBE-Umax-rest2 (window: 4–7) are recommended. Notably, TCBE-Umax-rest1 avoids editing at cytosine position 4, although it sacrifices some on-target efficiency. If no GG motif is detected, we suggest using editors with an ‘NGN’ PAM and testing TCBE-Umax-SpRY, TCBE-Umax-SpG and TCBE-Umax-NG to identify the most efficient editor. In the absence of an ‘NGN’ PAM, TCBE-Umax-SpRY is the preferred option, with PAM preference ranked as NAN > NYN (where Y is C or T), and the target cytosine should ideally fall within the primary editing window of the editor to maximize effectiveness. Overall, the TCBE-Umax effectors we have engineered hold substantial promise for overcoming the limitations of earlier base editors, enhancing disease model development and advancing potential gene therapy applications.
Fig. 7: Approach for selecting the optimal TadA-based CBE.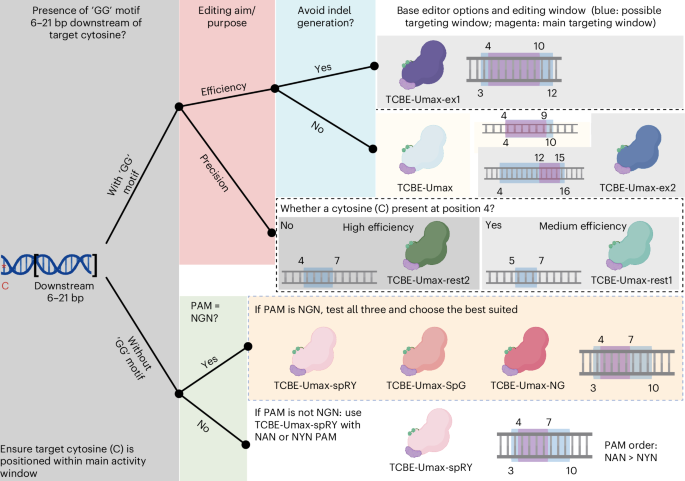
The selection of the right tool for C-to-T editing depends on the presence of a specific DNA sequence motif, and a suitable editor can be identified as follows: Identify GG motif within 6–21 base pairs downstream of the target cytosine (red asterisks). Option 1: If a GG motif is present, (1) for high efficiency, consider TCBE-Umax (window 4–10, with the PAM defined as positions 21–23), TCBE-Umax-ex1 (window 3–12) or TCBE-Umax-ex2 (window 4–16); if TCBE-Umax shows suboptimal activity at a given site, TCBE-Umax2 may be considered as an alternative to improve editing efficiency. (2) for high precision, choose TCBE-Umax-rest1 (window 5–7) or TCBE-Umax-rest2 (window 4–7). To minimize interference at the C4 position, TCBE-Umax-rest1 is the preferred choice, albeit with a slight trade-off in on-target efficiency. Option 2: If no GG motif is found, first test NGN PAM across all three editors (TCBE-Umax-SpRY, TCBE-Umax-SpG and TCBE-Umax-NG) and select the optimal one. If unavailable, use TCBE-Umax-SpRY, prioritizing PAM sequences in this order: NAN > NYN. (3) The edited cytosine should ideally be positioned within the main activity window to fully leverage the capabilities of each editor. The schematics, represented in different colours, illustrate various cytosine-based editing tools. The blue box represents the possible targeting window, while the magenta box indicates the main targeting window. Figure created in BioRender. Varshney, G. (2025) https://BioRender.com/3bcn8oc.