
Animals
Mice were male C57BL/6J mice (000664) and male Vglut1-cre mice (023527) mice obtained from The Jackson Laboratory (Bar Harbor) and male Glt1-G-CaMP740 mice obtained from RIKEN BioResource Research Center (G7NG817, RBRC09650). Mice were at least 8 weeks old at the time of surgery, before which they were group-housed in a temperature (72 ± 5 °F) and humidity (45 ± 15%) controlled vivarium under a 12 h–12 h light–dark cycle (lights on 06:00). After surgery, the mice were single housed to prevent cage mates from damaging intracranial implants. Experimental procedures were approved by the NIAAA and National Institute of Natural Sciences Animal Care and Use Committees and followed the NIH guidelines outlined in ‘Using Animals in Intramural Research’ and the local Animal Care and Use Committees.
General surgical and histological proceduresSurgery
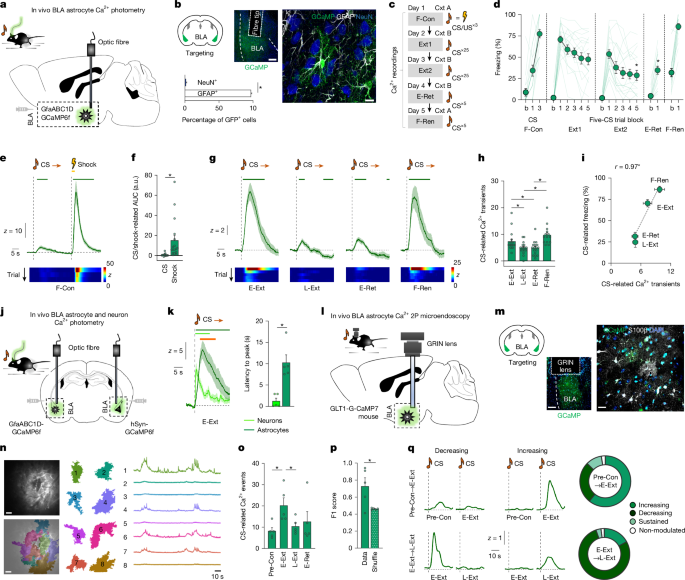
To target injection of viral constructs, optic fibres and GRIN lenses, mice were placed in a stereotaxic alignment system (Kopf Instruments) under isoflurane anaesthesia. Unless stated otherwise, viral constructs (see the relevant sections for details) were unilaterally (one-photon imaging, fibre photometry) or bilaterally (behavioural experiments, single-unit recordings) infused into the BLA in a volume of 0.36 µl over 10 min using a pulled-glass capillary (Drummond Scientific, 2-000-001; tip diameter, ~20 μm) connected to a Nanoject syringe (Drummond Scientific) at the coordinates: anteroposterior (AP) +1.42, mediolateral (ML) +3.27, dorsoventral (DV) −5.15, −4.95 and −4.75 (1 × 0.12 µl injection at each of the three DV coordinates) relative to bregma. After each injection, the glass capillary was left in place for 10 min to ensure diffusion. Testing began no sooner than 4 weeks after surgery, to allow for recovery and virus expression.
For behavioural experiments, the scalp wound was closed with tissue adhesive (GLUture) and a layer of topical antibiotic ointment applied. For fibre photometry experiments, optic fibres (MFC-400/430-0.66-6mm_SM3-FLT, B280-4653.6, Doric Lenses) were unilaterally implanted into the BLA at the coordinates AP +1.40, ML +3.25, DV −4.8 relative to bregma, and affixed to the skull with screws and acrylic dental cement (Coralite Dental Products). For one-photon and two-photon Ca2+ imaging experiments, a GRIN lens (ProView GRIN lenses; diameter, 0.6 mm; length, 7.3 mm; 1050-005442, Inscopix) was unilaterally implanted into the BLA at the coordinates AP +1.40, ML +3.25, DV −4.8 relative to bregma, and affixed to the skull with C&B-Metabond Quick Adhesive Cement System (Parkell). For single-unit recordings, a 16-tungsten microelectrode array (35 µm diameter, 150 µm spacing, configured in a semi-circle surrounding the tip of an optic fibre, Innovative Neurophysiology) was unilaterally implanted into the BLA, with the centre of the array at the coordinates: AP −1.50 mm, ML ±3.20 mm, DV −4.95 mm, relative to bregma. Moreover, a ferrule-fibre assembly (Thorlabs) was implanted at a 30° angle, such that the tip was positioned 0.5 mm above the microelectrode tips, with the array and optic fibre affixed to the skull with screws and dental cement (Coralite Dental Products).
Histology
On completion of testing, animals were terminally anaesthetized with sodium pentobarbital (50–60 mg per kg) and transcardially perfused with ice-cold PBS followed by ice-cold 4% paraformaldehyde in phosphate buffer. Brains were removed and suspended in 4% PFA overnight and then 4 °C 0.1 M PB for 1–2 days. For fibre photometry and imaging, the head was suspended in 4% PFA overnight before brain extraction. Coronal sections (50 μm) were cut with a vibratome (Leica VT1000 S, Leica Biosystems) in 0.1 M phosphate buffer and/or stored in 2% sodium azide in PBS, then cover slipped with mowiol-based mounting medium (17951, Polysciences) with Hoechst (1 μg ml−1, Thermo Fisher Scientific).
Unless stated otherwise, BLA containing sections were imaged at ×5 (HCX PL FLUOTAR objective, ×5/0.15) using either a Leica slide scanner with an ANDOR Zyla Monochrome camera and Leica DM6 motorized platform controlled by Aperio VERSA software (Leica Biosystems), an Olympus BX41 fluorescence microscope (Olympus America) or a laser-scanning confocal microscope (LSM700, Carl Zeiss) at ×20 (Plan-Apochromat air objective ×20/0.8 NA) and ×63 (Plan-Apochromat oil objective ×63/1.4 NA). Mice with absent/mistargeted viral expression, fibre optic or GRIN lens placement were removed from the analyses. Exceptions to these general histological procedures are described in the relevant sections below.
Immunohistochemistry
To verify the specificity of the astrocyte-targeting GCaMP6f, hM3Dq, hM4Di and CalEx viruses, immunostaining for astrocytic (Glial Fibrillary Acidic Protein, S100β) and neuronal (NeuN) markers was conducted, enhanced with primary antibodies to green fluorescent protein (GFP), TdTomato or DsRed. Free-floating sections were washed three times in PBS for 10 min then incubated in blocking solution (0.2% Triton X-100 and 5% normal goat serum in PBS) for 1 h at room temperature. The slices were then incubated overnight at 4 °C with primary antibodies in PBS containing 0.2% Triton X-100 and 1% normal goat serum. The antibodies used were: 1:1,000 chicken anti-GFP (ab13970, Abcam), 1:1000 rat anti-TdTom (EST203, Kerafast), 1:1,000 rabbit anti-DsRed (632496, Clontech), 1:1,000 guinea pig anti-GFAP (173400, Synaptic Systems), 1:100 rabbit anti-S100β (ab52642, Abcam) and 1:1000 rabbit anti-NeuN (ab104225, Abcam).
The next day, the sections were rinsed and incubated for 2 h at room temperature in a cocktail of secondary antibodies (1:500, goat anti-rabbit Alexa 405, A31556; 1:500, goat anti-chicken Alexa 488, A11039; 1:500, goat anti-rabbit Alexa 488, A11034; 1:500, goat anti-guinea pig Alexa 555, A21435; 1:500, goat anti-rat Alexa 555, A21434; 1:500, goat anti-guinea pig Alexa 647, A21450; 1:500, goat anti-rabbit Alexa 680, A21076; all antibodies from Life Technologies) in PBS containing 10% blocking buffer and 0.3% Triton X-100 solution. The sections were then rerinsed, mounted and coverslipped with mowiol-based mounting medium (17951, Polysciences).
BLA containing sections were imaged using a laser-scanning confocal microscope (LSM700, Carl Zeiss) at ×20 (Plan-Apochromat air objective ×20/0.8 NA). For each triple-staining, 2–3 BLA images were acquired per mouse. GFAP-positive cells and NeuN-positive cells, overlapping with virus expression were manually counted by experimenters blinded to experimental group using Fiji software (https://imagej.net/Fiji). The number of GFAP-positive astrocytes and NeuN-positive neurons that were also either GFP- or TdTomato-positive (that is, virus positive) were expressed as the percentage of total virus-expressing cells per 0.01 mm2 area.
Behavioural testingFear conditioning and extinction
Conditioning and extinction apparatus
For cued fear conditioning and extinction51,68,69,70, stimulus presentation was controlled by the Med Associates VideoFreeze system (Med Associates). Conditioning was conducted in a 27 × 27 × 11 cm chamber (80 lux) with opaque metallic walls and a metal rod floor that was cleaned between mice with a 79% water/20% ethanol/1% vanilla-extract solution to provide a distinctive odour. Extinction training and retrieval testing was conducted in a 27 × 27 × 14 cm chamber (20 lux) with transparent walls and a floor covered with wooden chips, sanitized with a solution of 99% water:1% acetic acid.
Conditioning entailed a 180 s baseline period followed by three presentations (60–90 s variable inter-CS interval) of a 30 s, 75 dB, white noise (CS) that co-terminated with a 2 s, 0.4 mA (for chemogenetic experiments, to offset the effect of pre-test injection-stress) or 0.6 mA (all other experiments) scrambled footshock US. There was a 120 s no-stimulus period after the final pairing before the mouse was returned to the home cage.
Extinction training began the day after conditioning, in a single 50× CS session or two 25× CS daily sessions (fibre photometry), bookended by 180 s pre-CS and post-CS periods. Each CS presentation was 30 s in duration, with a 5 s inter-CS interval except for a 60 s inter-CS interval for fibre photometry and 30 s for one-photon Ca2+ imaging to ensure CS-related Ca2 responses returned to baseline between CS-trials.
Extinction retrieval was tested the day after extinction training using the same procedure and context as extinction training, but with only 5× CS presentations.
Fear renewal was tested 1 day later in the same manner as extinction retrieval, but with testing conducted in the conditioning context.
Unless otherwise stated (seethe ‘In vivo fibre photometry’ and ‘In vivo one-photon neuronal Ca2+ imaging’ sections), freezing was scored manually every 5 s as no visible movement except that required for breathing71, and converted to a percentage ((number of freezing observations/total number of observations) × 100).
Fibre photometry behavioural control experiments
Three behavioural control experiments were conducted for fibre photometry. In US-no-CS and CS-no-US experiments, fear conditioning was tested as described above with the exception that, respectively, the US or CS was omitted. There was then a 5× CS test session the next day. In a no-extinction experiment, fear conditioning, extinction training and retrieval was tested as above, with the exception there were only 5× CS presentations at the start of the first extinction training session and another 5× CS presentations at the end of the second training session.
Anxiety-related behaviours
Novel open field
The novel open field72 was a 39 × 39 × 35 cm white Plexiglas square arena (centre of the arena illuminated to around 95 lux). The mouse was placed in a corner and allowed to freely explore the apparatus for 10 min. Total distance travelled and percent time spent in a 20 × 20 cm centre square was measured using the Ethovision automated tracking system (Noldus Information Technology).
Elevated plus-maze
The elevated plus-maze73 was ABS plastic (San Diego Instruments), consisting of 2 × 30 × 5 cm open arms (illuminated to around 95 lux) and two 30 × 5 × 15 cm closed arms (illuminated to around 20 lux) extending from a 5 cm2 central square and elevated 20 cm from the ground. The mouse was placed in the centre square to begin a 5 min test. The percentage of time spent in the open arms and total (open + closed) arm entries was measured by the Ethovision automated tracking system (Noldus Information Technology).
Light/dark exploration test
The light/dark exploration74 test apparatus comprised an opaque 39 × 13 × 16 cm black-Plexiglas compartment with a 13 × 8 cm aperture at floor level that allowed access to a larger, 39 × 39 × 35 cm, white-walled square Plexiglas arena (around 95 lux). The mouse was placed at the aperture (facing the opaque compartment) and allowed to explore the apparatus for 10 min. The percentage of time in the dark compartment and the total distance travelled throughout the apparatus was measured by the Ethovision video tracking system (Noldus Information Technology).
In vivo fibre photometrySurgery
A viral construct containing the genetically engineered Ca2+ indicators AAV5-pZac2.1 gfaABC1D-cyto-GCaMP6f (targeting the astrocyte cytosol75) (titre, 1.8 × 1013 vg per ml, Addgene plasmid 52925; provided by B. Khakh), AAV5-pZac2.1 gfaABC1D-lck-GCaMP6f (targeting astrocyte processes76) (titre, 1.8 × 1013 vg per ml, Addgene plasmid 52924, provided by B. Khakh) or AAV9-hSyn-jGCaMP7f-WPRE (targeting neurons77) (titre 2.5 × 1013 vg per ml, Addgene plasmid 104488, provided by D. Kim) was unilaterally injected (0.45 µl) into the BLA, and BLA-targeting optic fibres were unilaterally implanted, as described above in the ‘General surgical and histological procedures’ section. The same procedure was used for simultaneous photometry of astrocytes and neurons, but with AAV5-pZac2.1 gfaABC1D-cyto-GCaMP6f injected into the BLA of one hemisphere and pAAV.Syn.GCaMP6f.WPRE.SV40 (titre, 2.1 × 1013 vg per ml, Addgene plasmid 100837; provided by D. Kim) injected into the BLA of the other hemisphere (counterbalancing which hemisphere received each virus). Before behavioural testing, mice were handled for 2 min per day for 2 days and habituated to being connected to the optic fibre cables in the home cage for 1 h per day for 3 days.
Data collection
To record fluorescence signals via fibre photometry69,78, a RZ5P Processor acquisition system (Tucker-Davis Technologies) used two continuous sinusoidally modulated LEDs (Thorlabs) at 473 nm (511 Hz) and 405 nm (211 Hz) as a light source to respectively excite GCaMP and an isosbestic autofluorescence signal. The LEDs were connected to a mini cube (Doric Lenses) and each band-pass was filtered before being coupled to a single large core (400 μm), high-NA (0.48) optical fibre patch cord. Emitted light was projected through the same mini cube, passed through a GFP emission bandpass filter (500–525 nm) and focused onto a Newport Visible Femtowatt Photoreceiver (Doric Lenses). Light intensity at the tip of the patch cable (interface between patch cable and fibre implant) was in the 50–100 µW range for each channel. Photometry signals were temporally aligned to US and CS-onset using the Med Associates VideoFreeze system (Med Associates).
Data analysis
US- and CS-related changes in GCaMP activity were transformed into z scores of the 5-s period preceding each event (z = [ΔF − mean(ΔF(t = −5 to 0))]/s.d., where s.d. is the standard deviation of ΔF values during the pre-event period). Periods (bin size = 10 ms) in which peri-event activity differed from null were statistically determined as 95% confidence intervals (bCI) calculated from a 1,000-fold bootstrap estimate, as previously described79 (https://github.com/philjrdb/FibPhotom). Values for AUC were calculated using MATLAB’s built-in trapz function, which uses trapezoidal numerical integration to calculate the AUC (z score) between inputted x values (time) on a graph of z score versus time. US- and CS-related changes were examined in data z scored to the 5 s pre-stimulus period. The number of Ca2+ transients was calculated using a peak detection code (https://gist.github.com/antiface/7177333), with each recording visually inspected to ensure the accuracy of the code and the threshold parameter accordingly adjusted to differentiate between peaks and depressions (local maxima and minima) in the input signal. The results were expressed as the mean number of events or cumulative event frequency for a given experimental period and condition. CNO effects on transients were examined in data z scored to the 30 min pre-CNO period.
To examine the relationship between astrocyte Ca2 activity and movement during E-Ext and L-Ext, video recorded behaviour was analysed using the EzTrack software program80. ΔF/F Ca2+ values were aligned to instances of movement onset and offset as previously described23 and normalized to the session-average ΔF/F Ca2+ value.
In vivo two-photon Ca2+ imagingSurgery
A GRIN lens was unilaterally implanted into the BLA of Glt1-G-CaMP7 mice, as described above in the ‘General surgical and histological procedures’ section.
Data collection
Data were acquired using a Nikon A1R MP+ multiphoton laser-scanning microscope (Nikon Instech) controlled by NIS-Elements software (Nikon Instech). During imaging81, the mice were head-fixed but, to reduce the stress of immobilization, were able to ambulate on a rotating stainless-steel disc connected to a footshock generator (SG-1000S, Melquest). US and CS presentation was controlled by the Bpod HiFi Module HD State Machine r0.5 (Sanworks) and aligned to imaging frames via a multifunction I/O device (USB6343, National Instruments) running a custom script written in LabVIEW. A laser (Ti:sapphire, λ = 920 nm, Mai Tai DeepSee; Spectra-Physics) used as a light source to excite GCaMP was controlled by a galvanometer scanning an xy plane (512 × 512 pixels) at 500 ms frames using a water immersion ×16 objective (NA 0.8; Nikon). Fear conditioning and extinction was conducted as described above for fibre photometry, except for the omission of fear renewal and the addition of a pre-conditioning (Pre-Con) habituation session entailing 5× CS presentations, conducted the day before fear conditioning.
Data analysis
Images were analysed using ImageJ (v.1.37, National Institutes of Health) and MATLAB. Images were corrected for focal/xy plane displacements using StackReg and TurboReg and open-source code for 3D two-photon imaging registration (https://github.com/atakehiro/TurboReg_macro). Relative changes in Ca2+ fluorescence F were calculated by ∆F/F0 = (F − F0)/F0 (where F0 is a 25-frame moving average) using Astrocyte Quantitative Analysis software (AQuA)38. Overall Ca2+ changes across testing phases in AUC ΔF/F0 GCaMP values within regions of interest (ROIs) were identified using AQuA and custom MATLAB code from data concatenated across all five testing phases. Changes in Ca2+ event number were identified using AQuA and custom MATLAB code applied to individual testing phases. 11 ROIs with ΔF/F0 values >7 or a single AUC duration longer than 60 frames (2 fps) were excluded from the analyses. To assess plasticity across test stages, Ca2+ activity around a 20-s peri-CS period was used to determine CS-modulated astrocyte events (post-CS > pre-CS comparison using paired t-test (alpha, P < 0.05) on a given test stage and then segregate events into those exhibiting increasing, decreasing, sustained or consistent non-responsivity to the CS as a function of conditioning (Pre-Con to early extinction in cross test stage co-registered events) or extinction (early to late extinction).
CS decoding
A response vector was created by demarking the all-stage concatenated data according to presentation of the CS around a 5-s pre-CS period and 30-s post-CS period and decoded using a logistic regression model with an L2 penalty. To equate the length of the pre- and post-CS periods for decoding, a 5-s window from the post-CS period was constructed from activity in 10× non-contiguous 0.5-s intervals randomly sampled from the 30-s window. This construction process was repeated 1,000 times and the decoder performance was estimated by the mean average of the resultant F1 scores on held-out trials (trial-grouped fivefold cross-validation). Values were compared to the mean F1 scores obtained from circularly rotating the data (shuffle) 1,000 times with respect to CS presentation.
Histology
Histology was performed as described above in the ‘General surgical and histological procedures’ section.
Astrocyte manipulationshPMCA2w/b Ca2+ extruder (CalEx)
Surgery and behavioural testing
A viral construct containing the plasma membrane Ca2+ pump, CalEx82 (AAV5-pZac2.1-GfaABC1D-mCherry-hPMCA2w/b, titre 2.0 × 1013 vg per ml, Addgene plasmid 111568, provided by B. Khakh) or a tdTomato-expressing control virus (AAV5-pZac2.1 gfaABC1D-tdTomato, titre 7.0 × 1012 vg per ml, Addgene plasmid 44332, provided by B. Khakh) was bilaterally injected (0.36 µl per hemisphere) into the BLA, as described above in the ‘General surgical and histological procedures’ section. For simultaneous fibre photometry, AAV5-pZac2.1 gfaABC1D-cyto-GCaMP6f was unilaterally injected into the BLA, and BLA-targeting optic fibres were unilaterally implanted, as described above in the ‘In vivo fibre photometry’ and ‘General surgical and histological procedures’ sections. Before behavioural testing, mice were handled for 2 min per day for 3 days.
Chemogenetics
Surgery
A viral construct containing either hM3Dq (AAV5-GFAP-hM3D(Gq)-mCherry, titre 2.0 × 1013 vg per ml, Addgene plasmid 50478, provided by B. Roth), hM4Di (AAV-GFAP-hM4D(Gi)-mCherry, titre 1.0 × 1013 vg per ml, Addgene plasmid 50479, provided by B. Roth) or mCherry (AAV5-GFAP104-mCherry, titre 1.7 × 1013 vg per ml, Addgene plasmid 58909, provided by E. Boyden), was bilaterally injected into the BLA (0.36 µl per hemisphere), as described above in the ‘General surgical and histological procedures’ section. For simultaneous fibre photometry, AAV5-pZac2.1 gfaABC1D-cyto-GCaMP6f was unilaterally injected into the BLA and BLA-targeting optic fibres were unilaterally implanted, as described above in the ‘In vivo fibre photometry’ and ‘General surgical and histological procedures’ sections. Before behavioural testing, mice were handled for 2 min per day for 2 days and then injected with 0.1 ml of sterile saline intraperitoneally for 3 days to habituate to injection stress.
For DREADD actuation, CNO was injected intraperitoneally at a dose of 3.0 mg ml−1 per kg body weight, except in dose-comparison experiments in which CNO was injected at a dose 0.1 mg ml−1 per kg body weight. In a separate experiment to examine behavioural effects of a lower concentration hM3Dq virus, CNO was injected at a dose of 3 mg per kg in animals expressing a 1:8 diluted concentration of the hM3Dq-expressing virus (titre, 2.5 × 1012 vg per ml).
Behavioural testing
CNO was injected in separate groups of hM3Dq-expressing animals either (1) 30 min before fear conditioning; (2) immediately after fear conditioning; (3) 30 min before extinction training; or (4) 30 min before E-Ret. CNO was injected in hM4Di-expressing animals 30 min before extinction or 30 min before E-Ret. Ca2+ activity was measured in hM3Dq- and hM4Di-expressing animals, using in vivo fibre photometry, in the home cage for 30 min before CNO injection and then for the 30-min post-injection period up to the beginning of behavioural testing, as described above in the ‘In vivo fibre photometry’ section. After completion of behavioural testing, Ca2+ activity was recorded for 2 h after injection in a subset of mCherry-, hM3Dq- and hM4Di-expressing mice. For anxiety-related behaviour tests, CNO was injected into hM3Dq- and hM4Di-expressing animals 30 min before testing, with at least a 1-week interval between tests.
Chemogenetic control experiments
In an experiment to test for potential behavioural effects of CNO per se, mice bilaterally expressing a control virus in the BLA were injected before extinction with either vehicle or CNO. In an experiment to test for potential behavioural effects of hM3Dq expression per se, mice bilaterally expressing a hM3Dq or control virus in the BLA were injected before extinction with vehicle. A replicate experiment to match the design of the one-photon neuronal imaging (that is, hM3Dq actuation before F-Ret) was conducted in the same manner: that is, vehicle or CNO was injected before F-Ret in mice bilaterally expressing a hM3Dq or control virus in the BLA. Finally, the effects of hM3Dq actuation on shock-related freezing and flinching (the magnitude of response was manually scored on a scale of 0–5) were tested 30 min after CNO injection by presentation of five 0.4 mA footshocks each separated by a 60 s interval. Moreover, shock-related Ca2+ activity was measured during five 0.4 mA footshocks or, in a separate experiment, five 1.0 mA footshocks, as described above in the ‘In vivo fibre photometry’ section.
Electron microscopyFixation and immunohistochemistry
Mice were transcardially perfused with 0.1% glutaraldehyde/4% paraformaldehyde in 0.1 M phosphate buffer, pH 7.4, rinsed in the same buffer. Sections (40 μm) were cut with a vibratome (Leica VT1000 S, Leica Biosystems) and those containing BLA were rinsed in PBS consisting of 0.01 M phosphate buffer at pH 7.4 with 154 mM sodium chloride. The sections were then blocked for 15 min in 0.1% sodium borohydride in PBS, rinsed in PBS and blocked for 1 h in 1% BSA (Jackson ImmunoResearch Labs). The sections were incubated overnight in mouse anti-GFP (A-11120, 1859591; 1:500, Invitrogen) in PBS with 1% BSA at room temperature, rinsed in PBS and incubated for 30 min in biotinylated goat anti-mouse antibody (31802, 1:200, Invitrogen).
After rinsing in PBS, the sections were incubated for 30 min with an avidin/biotin complex (ABC) peroxidase reagent (Vectastain Elite ABC Kit PK-6100, Vector Labs) according to the manufacturer’s instructions, rinsed in PBS and reacted for 5 min with 1 mM 3,3-diaminobenzidine and 0.0015% hydrogen peroxide in PBS. After rinsing, the sections were incubated in 1% hydrogen peroxide in PBS for 15 min to quench the remaining peroxidase, then a second round of labelling was performed as above with rabbit anti-DsRed primary antibody (632496, 1904182; 1:500, Takara Bio), goat anti-rabbit secondary antibody (A32731, 1:200, Invitrogen) and ABC. The second antibody was detected with the Vector VIP peroxidase substrate (Vector Labs).
Microscopy preparation
BLA-containing sections were post-fixed in 2.5% glutaraldehyde/4% paraformaldehyde in 0.1 M cacodylate buffer, pH 7.4, rinsed in the same buffer, fixed for 1 h in reduced osmium (1% osmium tetroxide with 1.5% potassium ferrocyanide), rinsed in buffer and fixed for 1 h in 1% osmium tetroxide. After buffer rinses, the sections were rinsed quickly in water then dehydrated in a series of ethanol dilutions in water (50%, 70%, 90% and 100%) containing 1.5% uranyl acetate. The samples were transferred into acetone, infiltrated with LX-112 resin (Ladd Research) in acetone, embedded in pure resin and cured for 48 h at 60 °C.
Serial sectioning and imaging
One block from each of two mice was trimmed to expose the medial half of the basal amygdala. The sections were cut at 45 nm on a Leica UC7 ultramicrotome (Leica Biosystems) and collected on pioloform-coated slot grids. Serial sections were also cut from one block. Sections were imaged at ×5,000 on a JEOL 1400 TEM at 120 kV with an AMT NanoSprint43 digital camera. For 3D reconstruction, 11 serial sections were imaged. Reconstruct software83 was used for registration and segmentation and 3DS Max software (Autodesk) was used for rendering.
Inhibition of synaptic transmission
A tetanus toxin virus was used to inhibit synaptic release in BLA principal neurons, as previously described84. A viral construct containing tetanus toxin light chain85 (AAV1-DIO-GFP:TeNT, titre 4.0 × 1012 vg per ml, provided by L. Zweifel), was bilaterally injected in the BLA (0.36 µl per hemisphere) of Vglut1-cre+ mice or Vglut1-cre− controls, as described above in the ‘General surgical and histological procedures’ section. Then, 4 weeks later, fear conditioning and extinction was conducted as described above.
In vitro slice neuronal electrophysiological recordingsSlice preparation
A viral construct containing hM3Dq (AAV5-GFAP-hM3D(Gq)-mCherry, titre 2.0 × 1013 vg per ml, Addgene plasmid 50478, provided by B. Roth) was bilaterally injected into the BLA (0.36 µl per hemisphere), as described above in the ‘General surgical and histological procedures’ section.
At least 4 weeks after surgery, brains were removed and sliced as previously described86. In brief, the mice were deeply anaesthetized using isoflurane and then intracardially perfused with an ice-cold N-methyl-D-glutamine based solution (NMDG) (92 mM NMDG, 2.5 mM KCl, 1.25 mM NaH2PO4, 10 mM MgSO4, 0.5 mM CaCl2, 30 mM NaHCO3, 20 mM glucose, 20 mM HEPES, 2 mM thiouera, 5 mM Na-ascorbate and 3 mM Na-pyruvate, pH 7.3–7.4, 300–310 mOsm and saturated with 95% O2, 5% CO2). Brains were removed and coronal slices (300 µm) cut in ice-cold NMDG-based solution using a VT1200 S vibratome (Leica Biosystems). Slices were then held in NMDG-based solution maintained at 35 °C for around 12 min before being transferred to a room-temperature HEPES artificial cerebrospinal fluid (ACSF) (92 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 2 mM MgSO4, 2 mM CaCl2, 30 mM NaHCO3, 25 mM glucose, 20 mM HEPES, 2 mM thiouera, 5 mM Na-ascorbate and 3 mM Na-pyruvate, 300–310 mOsm, and saturated with 95% O2 and 5% CO2).
Data collection
Slices were transferred from the modified HEPES ACSF to a standard ACSF (118 mM NaCl, 2.5 mM KCl, 26.2 mM NaHCO3, 1 mM NaH2PO4, 20 mM glucose, 2 mM MgCl2 and 2 mM CaCl2, at 25 °C, pH 7.4, 300–310 mOsm, and gassed with 95% O2 and 5% CO2). Neurons were visualized using an infrared differential interference contrast camera on a BX51WI system (Olympus). Whole-cell patch clamp recordings were performed using micropipettes pulled from a borosilicate glass capillary tube using a PMP102 Micropipette Puller (MicroData Instruments). Electrode tip resistance was 3–5 MΩ. Recordings were acquired using an Axon Multiclamp 700 and Axon Digidata 1550 A (Molecular Devices), sampled at 10 kHz, low-pass filtered at 3 kHz and analysed in Igor Pro 8 (WaveMetric). Access resistance was continuously monitored and changes greater than 20% from the initial value were excluded from the analysis. Recordings were made in 1–2 cells per mouse.
To assess the effects of hM3Dq actuation of BLA astrocytes on nearby neuronal activity, a potassium gluconate-based intracellular solution (155 mM K-gluconate, 4 mM KCl, 10 mM HEPES, 4 mM MgATP, 0.3 mM Na3GTP, 10 mM phosphocreatine, pH 7.3, 285–290 mOsm) was used. Neurons were first current clamped and the membrane potential was maintained at −70 mV while a 500 ms current step was applied every 5 s, starting at −200 pA to 600 pA increasing by 50 pA with every step. The neurons were then held in voltage clamp at −70 mV and spontaneous EPSCs (sEPSCs) measured. After a 5 min baseline period, 10 μM CNO was bath applied and maintained in CNO for 35 min and sEPSCs again measured as above.
In vivo one-photon neuronal Ca2+ imagingSurgery
A viral vector containing the Ca2+ indicator jGCaMP7f77 (pGP-AAV-syn-jGCaMP7f-WPRE, titre 2.5 × 1013 vg per ml, 1:5 dilution in Dulbecco’s PBS, Thermo Fisher Scientific, Addgene plasmid 104488, provided by D. Kim and the GENIE Project) was unilaterally infused (0.50 µl) into the BLA, as described above in the ‘General surgical and histological procedures’ section. Then, 3–4 weeks later, before behavioural testing, mice were handled for 2 min per day for 3 days and then habituated to being connected to the microscope and tether for 30 min per day for 3 days.
Data collection
Fear conditioning and extinction was conducted as described above, with 3 mg per kg CNO or vehicle injected into mice bilaterally expressing hM3Dq in BLA astrocytes 30 min before F-Ret in one experiment or, in a separate experiment, 30 min before extinction training. BLA neurons were imaged using a miniature microscope (nVistaTM 3.0, Inscopix) and IDAS HD software (Inscopix) at a frame rate of 20 Hz with an LED power of 10–60% (0.9–1.7 mW at the objective, 475 nm), analogue gain 1, 1,080 × 1,080 pixels. Timestamped imaging frames were collected for temporal alignment using the Med Associates VideoFreeze system (Med Associates).
Data analysis
For data preprocessing, imaging frames were downsampled to 10 Hz, spatially filtered and motion-corrected using Inscopix software, then putative neurons were identified using constrained non-negative matrix factorization (CNMF-E)54. Traces for each neuron were manually inspected to exclude false-positive or false-negative cell mask allocation. Raw Ca2+ traces were obtained by averaging pixel values in each mask. Slow drift of the baseline signal over the course of minutes was removed using a low-cut filter (Gaussian, cut-off 2–4 min). Relative changes in Ca2+ fluorescence F were calculated by ∆F/F0 = (F – F0)/F0 (where F0 is the median fluorescence of the entire session trace). Ca2+ activity data were analysed using the Python programming language (https://www.python.org/). To compute CS-related Ca2+ responses, time-normalized AUC fluorescence values (z scored to the entire session) were calculated for each stimulus presentation. To examine the relationship between astrocyte Ca2+ activity and freezing, video-recorded behaviour was analysed using the EzTrack software program, as described above in the ‘In vivo fibre photometry’ section.
CS-modulated activity
CS modulation was defined by calculating the delta in activity for the 5-s post-CS relative to the 5-s pre-CS period and a corresponding distribution of 500 null values obtained from activity circularly rotated with respect to CS presentation. Modulated neurons were statistically defined based on the proportion of null values greater than the observed value (P < 0.05). CS modulated neurons were statistically defined based on the proportion of null values greater than the observed value (P < 0.05) on a given test stage. These results were used to segregate cross test stage co-registered neurons into subsets exhibiting increasing, decreasing, sustained, or consistent non-responsivity to the CS as a function of fear conditioning (Pre-Con to F-Ret).
Neuronal topography
To determine the spatial organization of CS-modulated neurons within the field of view, the Euclidean distance (arbitrary units) between individual CS-related neurons and their respective centroid was calculated, as previously described54.
Decoding treatment group identity by animal
CNO treatment group identity was decoded using a logistic regression model with an L2 penalty (balancing the number of animals per group, n = 7) and creating a response vector based on specific features of F-Ret CS-related neuronal activity during a 10-s post-CS period demarked according to group. To ensure that the same number of neurons was used for each mouse, a randomly selected (through sampling with replacement) subset of n = 5 neurons was sampled 100 times and the average computed. The decoder performance was then evaluated using a specific neural feature (see below) from mean F1 scores across 14 cross-validation folds (that is, 1-fold per mouse) and compared to the mean F1 scores obtained from circularly rotating the data (shuffle) with respect to group identity, 100 times. The following features were examined: mean activity of positively CS-modulated, negatively CS-modulated and non-modulated neurons (as defined above), the s.d. of activity of all neurons during the 5-s post-CS period and the decomposed activity values of the three highest-eigenvalue principal components computed by PCA on z-scored, trial-averaged and mean-normalized activity from a 10-s peri-CS period.
Decoding CS as a function of population size
A response vector was created by demarking data according to presentation of the CS around a 10-s peri-CS period and decoded from the neuronal data using a logistic regression model with an L2 penalty (balancing the number of animals per group, n = 7). Decoding was conducted with the number of neurons available to the decoder (randomly selected by sampling without replacement) increasing in one-neuron increments from 2 to 100. The performance at each population size was estimated as the mean held-out F1 score of 50 decoding models, each trained on a random sample of neurons of the corresponding size and compared to values of 50 iterations of circularly rotated data (shuffle) with respect to CS presentation.
To model the relationship between population size and decoding performance, a saturating function50,87 was fit to the data. The saturating function was defined as \(y=bn/a+n\), where y is the decoder performance, n is the population size, b is the performance at an infinite number of neurons and a is the saturation rate. The saturating function was fit to F1 scores using the curve_fit function from the SciPy Python package. To estimate the variance of model fits, the function was fit to the data 1,000 times, each time with a randomly selected combination of the 50 model fits (50 random samples with replacement) per population size.
Relationship between freezing and CS presentation decoding
CS presentation was decoded from neuronal activity during F-Ret as described in the ‘Decoding CS as a function of population size’ section, but here using the entire population of neurons available (balancing the number of animals per group, n = 7). The resultant decoder coefficient values for each neuron were averaged by mouse and the mouse-averaged correlated with the respective F-Ret CS-related freezing values.
k-Means clustering
To identify neurons that were CS-responsive on extinction and differentially activated by CNO treatment, neuronal activity in the 5-s pre-CS through 30-s post-CS period during L-Ext and E-Ret was subject to k-means clustering (n = 4 clusters).
In vivo neuronal electrophysiological recordings and phototaggingSurgery
A viral vector containing CalEx was bilaterally infused into the BLA, as described above in the ‘hPMCA2w/b Ca2+ extruder’ and ‘General surgical and histological procedures’ sections. Additionally, the retrograde-travelling excitatory opsin, channelrhodopsin (retroAAV-hSyn-hChR2(H134R)-eYFP, titre 2.5 × 1013 vg per ml, Addgene plasmid 26973, provided by K. Deisseroth) was bilaterally infused (0.20 µl) into dmPFC at a 20° angle at the coordinates: AP +1.95 mm, ML ±1.00 mm, DV −1.90 mm, relative to bregma, as described above in the ‘General surgical and histological procedures’ section. During the same surgery, a microelectrode array and optic fibre were unilaterally implanted into the BLA88, as described above in the ‘General surgical and histological procedures’ section. Before testing, the mice were habituated to the recording tether and patch cable for 20 min in the home cage handled for 2 days. During habituation, 20 Hz blue light pulses (fifty 1 s trains of 20 Hz light pulses with a 3 s intertrial interval) were delivered through the optic fibre to identify light-responsive isolated single units. Light responsivity was reconfirmed after each behavioural test session.
Data collection
Fear conditioning, extinction training and retrieval were tested as described above with the exception that the CS consisted of a 20 s train of white noise pips89 (500 ms pips delivered at 1 Hz, 70 dB) and the extinction context was a clear acrylic, cylindrical 30 cm diameter chamber with an open top to accommodate the tether connecting the head-stage. Electrophysiological and behavioural videos were acquired using the Omniplex Neural Data Acquisition System and Cineplex Behavioural Research System (Plexon). Radiant Software (Plexon) was used to generate TTL pulses to control stimulus-presentation. Plex Control Software (Plexon) recorded TTL signals to synchronize electrophysiological recordings with stimulus-presentation and videos of behaviour.
Data analysis
Single units were sorted manually using Offline Sorter v.4.0 (Plexon) and analysed using NeuroExplorer v5 (Nex Technologies). To identify CS-responsive units, data during a 500 ms window following the onset of each pip during each of the 5 CS presentations of E-Ext, L-Ext and E-Ret were binned in 100 ms bins and z-score normalized to the 500 ms pre-pip baseline period, as follows: (CS bin value − baseline mean value)/(s.d. of the baseline mean value). Units with at least 1 of 5 bins with a value of >2.58 during the post-pip period were deemed significantly different from baseline (P < 0.01) and classified as pip-responsive. To identify freezing-related unit activity, firing rates during freezing epochs were compared to firing rates during movement epochs on the 5 CS presentations and associated ITI periods of E-Ext, as follows: (Hz rate during freeze − Hz rate during move)/(Hz rate during freeze + Hz rate during move).
Histology
To verify electrode placements, mice were anaesthetized with 2% isoflurane and marking lesions made by passing current (40 µA for 2 s) through each of the microelectrodes (S48 Stimulator and Model CCU1, Grass Technologies). At least 1 day later, mice were euthanized with 150 mg per kg Euthasol (Henry Schein) and transcardially perfused with 4% PFA solution. The brains were extracted and stored in PBS. Coronal sections (50 µm thick) were cut with a VT1000 vibratome (Leica), mounted on slides and cover slipped with Invitrogen Fluoromount-G mounting medium with DAPI. Viral and fibre placements and marking lesion sites were documented with the aid of a Keyence BZ-X microscope (Keyence).
Statistical analysis
Data were analysed using Python (https://www.python.org), MATLAB (MathWorks) and Prism (GraphPad) software. Group effects were examined using two-tailed unpaired or paired Student’s t-tests or ANOVA, depending on the number of independent variables. Post hoc tests (Šídák’s and Fisher’s LSD) were conducted after ANOVA. χ2 tests were used to analyse group proportion differences. Sample sizes were based on pilot and previous (similar types of) experiments and were not statistically predetermined. Experiments were powered to match sample sizes typical of the technique reported in the field, although no formal power analysis was performed a priori. Images shown were selected from multiple independent samples (that is, different animals). P < 0.05 was considered the threshold for statistical significance, and P < 0.0001 is the lowest P value reported. Experiments were biologically replicated across multi-animal batches (animal N and n numbers for each experiment are provided in the corresponding figure legends and Supplementary Table 1). Representative example micrographs depicted in Figs. 1b,m,n, 2b,h, 3b and 4b and Extended Data Figs. 1a,h, 3a,i, 4a, 5k, 9a,b, 10b, 11g and 12i) were selected from at least three independent animals, except for electron microscopy, which was based on two mice. Experimental groups were randomized, and experimenters were blinded to group allocation whenever possible.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.