
Newswise — Researchers from Peking Union Medical College and Chinese Academy of Medical Sciences have conducted a study entitled “Identification of novel mutations in EYA3 and EFTUD2 in a family with craniofacial microsomia: evidence of digenic inheritance”. This study was published in Frontiers of Medicine, Volume 17, Issue 5.
The study originated from a unique family: the proband was a 7-year-old Han Chinese girl who underwent auricle reconstruction surgery due to congenital microtia. Examinations revealed hypoplastic low-set left auricle, external auditory canal atresia leading to conductive hearing loss, along with mild facial asymmetry, mandibular retrognathia, and zygomatic flattening—indicating hypoplasia of the left maxilla and mandible. Her mother had a history of right-sided congenital microtia and had undergone auricle reconstruction. Family investigation showed no similar symptoms in other relatives, initially suggesting the disease might be transmitted in an autosomal dominant manner between the mother and daughter. However, phenotypic heterogeneity and incomplete penetrance pointed to the possibility of non-Mendelian inheritance.
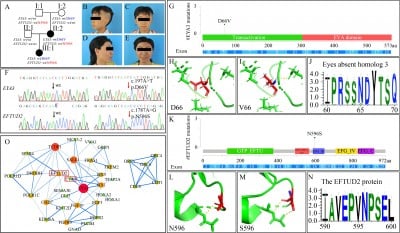
To identify the genetic cause, the research team performed whole-genome sequencing on three family members (the proband and her parents), with no suspicious structural variations detected. By screening missense mutations with a population frequency below 0.001 that co-segregated with the phenotype, and combining with the classification guidelines of the American College of Medical Genetics and Genomics (ACMG), two key genes—EYA3 and EFTUD2—were finally identified. Sanger sequencing verification showed that both mutations were present in the proband and her affected mother, but not in the unaffected father. Further testing of the unaffected maternal grandparents revealed that each carried one of the mutations.
The pathogenicity of the mutations was evaluated using multiple bioinformatics tools: REVEL scores indicated the pathogenicity prediction values of the EYA3 and EFTUD2 mutations were 0.733 and 0.643, respectively; VarCards database analysis showed that 19 out of 23 algorithms and 20 out of 23 algorithms predicted the two mutations as harmful, respectively; Sequence alignment across 46 species revealed that the amino acid residues at the mutation sites were highly conserved during evolution; Protein three-dimensional structures constructed using PyMOL software showed that the mutations caused changes in the polar contacts between amino acid residues and adjacent atoms. In addition, expression data from the Mouse Genome Informatics Database showed that both EYA3 and EFTUD2 were expressed in the first pharyngeal arch during embryonic days E9-E10.25, suggesting they have similar functions at specific spatiotemporal stages. The protein-protein interaction network constructed using the STRING database further indicated that the two genes might be associated in the craniofacial development-related network.
In terms of gene function: EYA3 is involved in post-injury repair and apoptosis regulation, and mutations in its paralogous gene EYA1 have been confirmed to be associated with diseases such as otic-renal syndrome. The EYA3 D66V mutation identified in this study is located in the N-terminal region and is partially conserved in evolution. Heterozygous mutations in EFTUD2 can cause mandibulofacial dysostosis with microcephaly. The EFTUD2 N596S mutation identified here is located in the conserved domain III of elongation factor G, and pathogenic mutations in the same region have been confirmed to cause exon skipping.
The research team analyzed three possible explanations for the coexistence of two pathogenic mutations in the family: typical digenic inheritance, “pseudo-digenic inheritance,” or independent segregation of the two mutations leading to different diseases. Combining features such as only microtia (without other abnormal phenotypes related to EFTUD2 mutations) in the mother and daughter, the study suggested that the family might involve digenic inheritance. However, due to the small family size and lack of direct functional data support, the relevant conclusions still require verification with more cases. Notably, the maternal grandparents, who carried only a single mutation each, showed no disease symptoms. The study hypothesized this might be related to the polygenic additive modification effect. No environmental risk factors such as gestational diabetes or drug use were reported in the family, indicating that genetic factors may play a major role.
Through genetic analysis of a three-generation family with craniofacial microsomia, this study reports the co-segregation of two novel mutations in EYA3 and EFTUD2, providing new case clues for the complex genetic mechanism of craniofacial microsomia. The study not only supports the possibility of digenic inheritance involved in the pathogenesis of this disease but also reveals the synergistic effect of related genes in craniofacial development through functional network analysis, which helps to deepen the understanding of the molecular basis of disease phenotypic variation and genetic heterogeneity.
For more detailed information, the full paper is available at: https://journal.hep.com.cn/fmd/EN/10.1007/s11684-023-1000-3.