
Though critical for gas exchange, the lung is also a barrier organ and as such represents a common site of injury. In addition to exposure to environmental hazards and toxins, it is also subject to a wide range of respiratory pathogens. While many such pathogens can be effectively cleared while restricted to the upper airways, others, including some strains of influenza and coronaviruses, can infect distal epithelial cells and drive overexuberant inflammatory responses, causing severe injury to the blood-gas barrier and resulting in acute respiratory distress syndrome, which continues to bear a high mortality rate of > 35% [1].
In response to such severe injury, the mammalian lung employs a number of distinct progenitor cell types capable of responding to injury. In the airways, basal cells expressing the transcription factor p63 are well described to act as a progenitor cell for other airway cell type including club, ciliated, goblet, neuroendocrine, and tuft cells [2,3,4,5,6,7]. In the alveolar compartment, which is anatomically and functionally distinct from conducting airways, normally quiescent alveolar type 2 cells can transiently adopt progenitor features, proliferating and differentiating to replace alveolar type 1 cells responsible for gas exchange. However, in severe injury scenarios, this distinction between the airways and alveoli appears to be disrupted, in that basal-like cells can migrate from the airways into the alveolar region, aiding in barrier restoration but ultimately contributing only in very small part to the restoration of AT1s and AT2s [8,9,10,11,12,13]. What’s more, these dysplastic regions of bronchiolized tissue appear to persist long term, at least 1 year in mouse models [14]. Identifying methods to promote transdifferentiation of basal cell-derived dysplastic tissue into more regionally appropriate alveolar cell types thus represents an important goal to aid in true, euplastic alveolar regeneration (i.e., complete restoration of normal form and function).
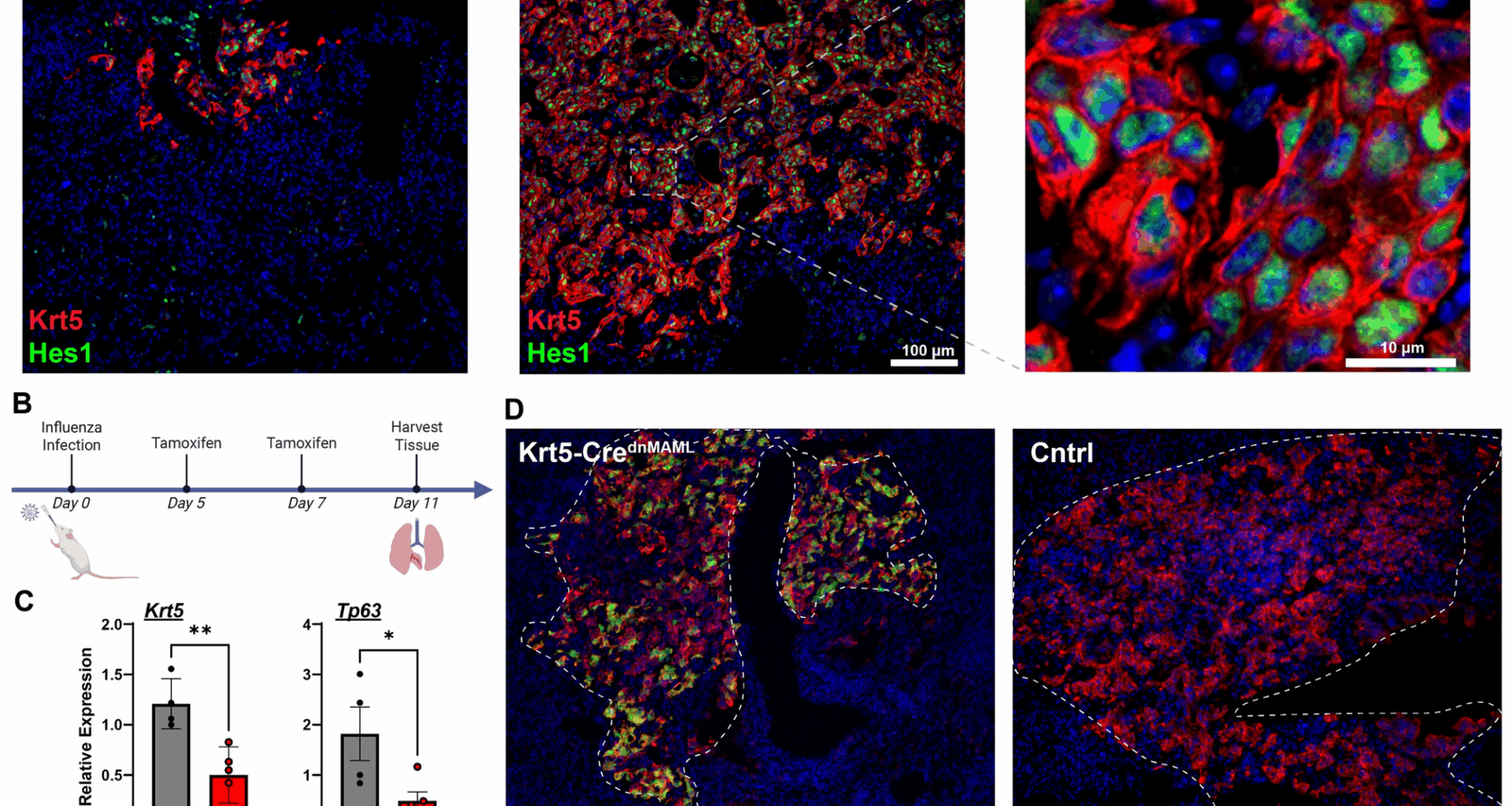
Several groups have identified signals involved in promoting dysplastic repair. For instance, HIF1a deletion or Wnt activation prior to injury blunts the dysplastic response in favor of greater contribution from distal airway secretory cells (club cells and BASCs) [9, 15] which appear quite capable of generating AT2s and AT1s [6, 16]. Upon observing strong Notch target gene expression (Hes1) in dysplastic regions of tissue, initial work from our group demonstrated that treatment with γ-secretase inhibitors, widely used to inhibit Notch signaling via preventing cleavage of the intracellular domain, early in influenza injury similarly blunted Krt5 + cell expansion [8]. Follow-up work has now demonstrated that much of this effect can be ascribed to paracrine crosstalk between activated fibroblasts, with dysplastic basal cells providing Jag2 ligands to Notch3 receptors on the fibroblasts, which in turn generate trophic signals for basal cell expansion [17]. Treatment of post-influenza mice at later stages with γ-secretase inhibitors, in conjunction with dexamethasone and IBMX, resulted in a slight increase in AT2-like differentiation from Krt5-CreERT2 traced cells [8]. However, in spite of the widespread use of γ-secretase inhibitors to experimentally probe the Notch signaling pathway, it is increasingly recognized that γ-secretase has a very large number of substrates aside from just the Notch receptors, including but not limited to Eph receptors, Wnt receptors like Lrp6, Cdh1, and Trop2 [18, 19]. As such, it is difficult to ascribe any effect of γ-secretase inhibition solely to antagonism of the Notch pathway.
With this information in mind, here we reexamined previous models utilizing a genetically targeted approach to block all Notch signaling via inducible expression of a dominant negative MAML protein (dnMAML) [20, 21]. This approach not only avoids the lack of specificity from γ-secretase inhibition but also has the advantage of reducing Notch signaling to the basal levels without the “derepression” phenomenon sometimes observed with RBPJ deletion [22]. As we recently reported, we observed that both dnMAML overexpression and treatment with the γ-secretase inhibitor DBZ significantly reduced expression of Notch target genes Hes1, Hey1, and Hes2, but only DBZ treatment exhibited significant growth and proliferation defects in vitro (see Supplemental Fig. 12, Jones et al. [17]). We therefore utilized these animals to re-examine the role of Notch signaling in injury-induced basal cell expansion and differentiation in vivo.