
Scientists have used a genome-editing technique called base editing to correct a mutation in a baby who had been diagnosed with a rare genetic disease. The bespoke therapy, in which the treatment was delivered through lipid nanoparticles, was developed in record time, as reported by Musunuru et al.1 in The New England Journal of Medicine.
Read the paper: Patient-Specific In Vivo Gene Editing to Treat a Rare Genetic Disease
Around 10,000 rare diseases are estimated to exist, affecting more than 300 million people globally2. However, when diseases that are ultra-rare (with a prevalence of less than one in 50,000 individuals) and hyper-rare (affecting less than one in 108 individuals) are also considered, these figures are underestimates, highlighting the profound burden of these illnesses both for health-care systems and in terms of human suffering. The rapid advance of nucleic-acid-based genetic therapies, supported by robust clinical validation, is providing opportunities for the development of tailored treatments and early intervention.
In the first example of a truly personalized genetic therapy, a molecule called an antisense oligonucleotide (ASO) was custom engineered to treat Mila, a girl with a rare neurogenerative condition called Batten disease3. Genetic testing identified a mutation in the gene MFSD8. Building on the clinical success of nusinersen, a drug approved by the US Food and Drug Administration (FDA) for spinal muscular atrophy4, researchers developed an ASO to restore MFSD8 expression. The resulting therapeutic, named milasen, led to a notable reduction in the duration and the frequency of the seizures that Mila experienced. This effort established a framework for the rapid design, synthesis and laboratory validation of individualized ASOs, demonstrating the technical and regulatory feasibility of individually tailored therapeutics. Treatments of this kind, which are designed to match the genetic make-up of a single individual, are often referred to as N = 1 or N-of-1 therapeutics.
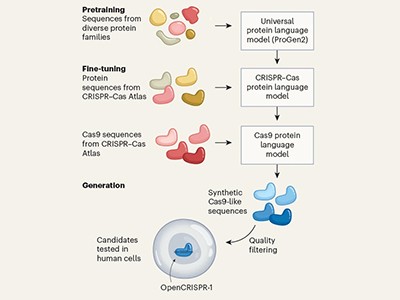
AI expands the repertoire of CRISPR-associated proteins for genome editing
Despite these early successes, broad implementation of genetic therapies has yet to be achieved. Only a limited number of individuals have received such treatments, and efforts to scale up their production face substantial scientific, regulatory and economic challenges. In response to these issues, various initiatives, such as the Rare Therapies Launchpad, the N=1 Collaborative (N1C) and the n-Lorem Foundation, are working to develop sustainable systems for wider implementation5−7.
The emergence of CRISPR–Cas genome-editing technologies to alter DNA directly is enabling targeted modifications of disease-driving variants of genes, with high specificity and efficiency. Clinical applications based on this idea have yielded durable beneficial therapeutic outcomes. Editing of a person’s own haematopoietic stem and progenitor cells (stem cells in the bone marrow that give rise to blood cells) has curative potential for blood disorders called β-haemoglobinopathies. Work in this area culminated in the clinical approval of a CRISPR-based therapeutic for sickle cell disease8. This treatment, marketed as Casgevy, has, in turn inspired the development of gene-editing therapies for other types of inherited blood disorders and genetic immunodeficiencies.
For diseases driven by genes expressed in the liver, delivery of genome-editing components inside lipid nanoparticles has produced sustained, targeted effects, favourable safety profiles and promising clinical outcomes9,10. Therapies being developed for the conditions transthyretin amyloidosis and hereditary angiooedema have progressed to clinical development10 (see go.nature.com/4mvjmg7; go.nature.com/4memswz and go.nature.com/3hzgpke). Furthermore, various CRISPR-editing approaches11, such as base-editing and prime-editing technologies, have shown clinical promise (see go.nature.com/4fjqxpp; go.nature.com/4tnjsqj and go.nature.com/45mu2gd), and other types of gene-editing system are in early-phase clinical trials.
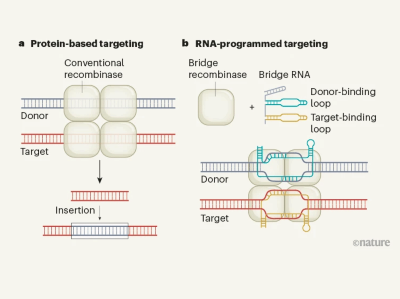
Programmable RNA-guided enzymes for next-generation genome editing
These advances have expanded the repertoire of clinically validated genome-editing systems that are capable of correcting a broad spectrum of disease-causing genetic mutations. This progress has boosted collaborations between academia and industry, such as the Danaher–Innovative Genomics Institute (IGI) Beacon for CRISPR Cures, which aims to accelerate the development and distribution of personalized genome-editing therapies in a sustainable, affordable and equitable manner12,13. In parallel, academic investigators are exploring streamlined pathways for rapid diagnosis and individualized design of therapeutics.
In early 2023, Musunuru and colleagues initiated a systematic effort to develop a method that enabled the identification and rapid correction of rare metabolic disorders. This culminated in the development and clinical implementation of the first personalized in vivo gene-editing therapy for a baby.
The baby in question, KJ Muldoon, was diagnosed shortly after birth with a severe form of a disorder caused by a mutation in the gene CPS1. This inherited condition affects the urea cycle and causes a build-up of ammonia, a toxic product of protein breakdown, leading to brain and organ dysfunction. Although the condition can be managed by interventions such as limiting protein intake, and using nitrogen-scavenger medications, in severe cases — for example, when the enzyme encoded by CPS1 is not produced at all — high levels of ammonia can cause irreversible brain damage, coma and death.
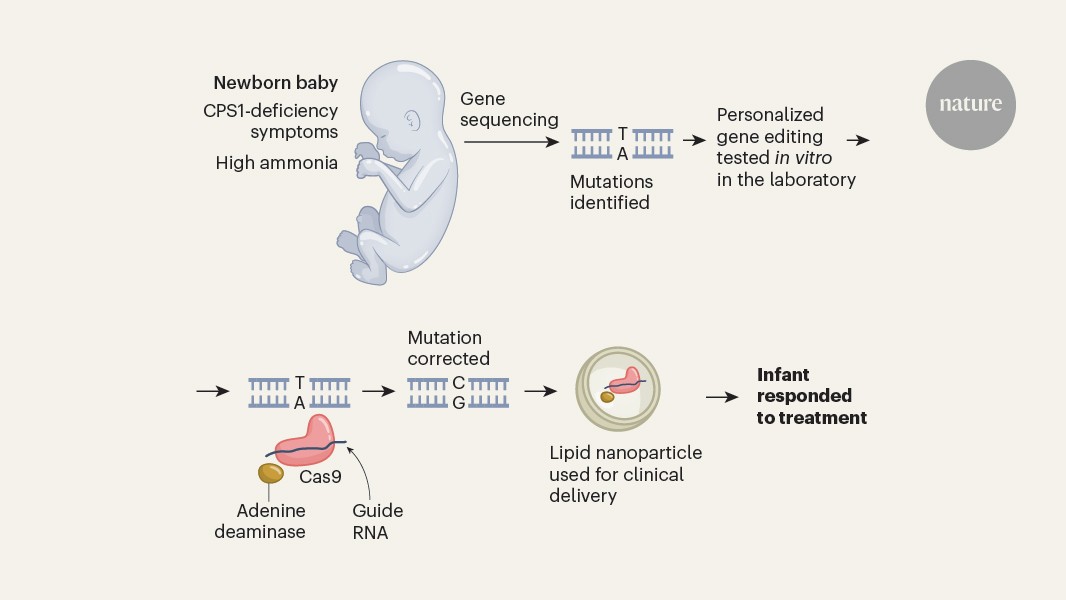
Musunuru et al. used rapid genomic sequencing to identify maternally and paternally inherited mutations in the CPS1 gene. The authors determined that the paternally inherited version of the mutation was amenable to correction by a tool called an adenine base editor, which could replace the mutant adenine base with a guanine base (Fig. 1).
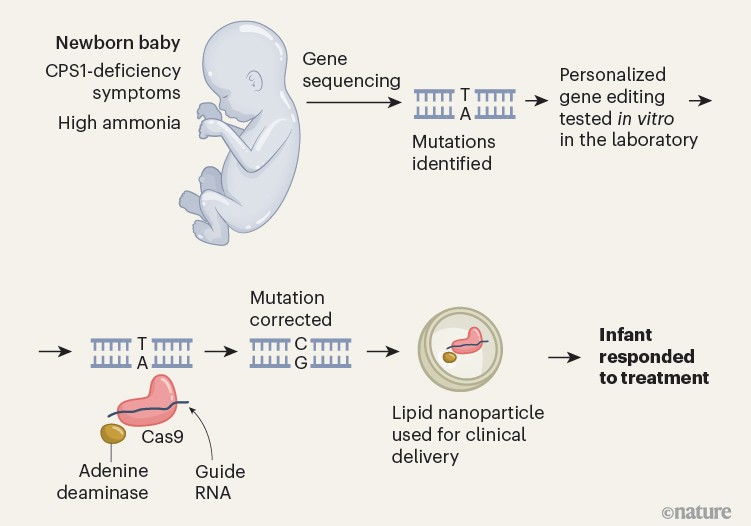
Figure 1 | A gene-editing therapy for an infant. Musunuru et al.1 report a therapy used to treat a baby who had symptoms of a metabolic disease associated with inherited mutations in the gene CPS1. Symptoms included the presence of high levels of ammonia. The disease was diagnosed by genomic sequencing of the newborn, which revealed a mutation that generated an adenine (A) nucleotide (which pairs with a thymine (T) nucleotide) instead of a guanine (G) nucleotide (which pairs with a cytosine (C) nucleotide). The authors used a gene-editing method (harnessing the protein Cas9, a guide RNA and the enzyme adenine deaminase) that was capable of changing an adenine nucleotide to a guanine nucleotide, and verified that this corrected the mutation when tested in vitro. The therapy was delivered to the infant through a lipid nanoparticle, and the child’s symptoms improved in response to the treatment.
Musunuru et al. assembled an academia–industry collaboration to generate, test and deliver the therapeutic in just over six months. The authors developed in vitro cell lines and mouse models engineered to express the same variants of the CPS1 gene that KJ had. Musunuru and colleagues then screened various adenine base editors and selected a base editor and a guide RNA sequence that efficiently corrected one of the CPS1 mutations, restoring gene expression and enzymatic activity in mice that received the treatment. To assess whether there was any risk of off-target gene editing (in which DNA sites other than the one intended are modified), personalized off-target analysis was performed using KJ’s whole genome sequence.
Industry partners Danaher and Acuitas Therapeutics supported the study and manufactured the clinical-grade treatment, which was named kayjayguran abengcemeran (K-abe) in honour of its recipient. Throughout this process, the team engaged with staff from the FDA, who provided timely feedback on matters relating to laboratory testing and therapeutic development, including a toxicology study in non-human primates. After a one-week review of the team’s application to test the treatment as an investigational new drug, the FDA cleared it for a clinical trial with institutional oversight.
KJ’s parents provided written informed consent, and he received three escalating doses of the treatment (see Nature https://doi.org/g9kbgg; 2025), beginning just before he reached seven months of age. The therapy was well tolerated, with no serious adverse events. Clinical improvements were seen after the first dose, including increased tolerance to dietary protein, with further gains after the second dose. Although it did not provide a cure, the intervention transformed a severe disease that would normally require a liver transplant in the early months of life into a condition that can be managed more easily, as assessed in this initial follow-up.
This result underscores the feasibility of integrating rapid genetic diagnosis with individualized therapeutic design in a time frame that works for clinical intervention. However, it also raises key questions about access and sustainability. Although the technological foundation for individualized genomic medicines now exists, the reliance on philanthropic financial support — as was the case for the development of this treatment — is unlikely to enable widespread implementation.
To support the sustainable and equitable development of individualized genomic medicines, several key elements must converge. These include the establishment of regulatory platform-based approval pathways to streamline the evaluation of therapies across diseases14; the development of funding models that involve partnerships between public and private entities; and investment in a shared private−public infrastructure and knowledge base to accelerate progress. Finally, integrating genomic sequencing into newborn screening programmes could enable early identification and timely intervention for treatable genetic disorders, maximizing the clinical impact of these emerging therapies.