
A previously healthy 14-year-old girl exhibited symptoms of severe vomiting for 12 days, fever and cough for 5 days and epileptic seizure for 3 h. She was admitted to the Hainan Affiliated Hospital of Hainan Medical University on 13 March 2021. She had no remarkable medical or drug history nor any recent history of vaccination. On admission, the neurological examinations revealed disturbance of consciousness (Glasgow Coma Score 12), paralytic paraplegia of both lower limbs, urinary incontinence, nystagmus and hiccups. The brain magnetic resonance imaging (MRI) from the previous hospital revealed hyperintensity in the SCC alone (Fig. 1). Wet rales were audible in both lungs. Chest computed tomography revealed an infiltrate in the right upper lung. On the first day of hospitalisation, the disease rapidly progressed, and the patient had sudden difficulty breathing, with blood gas analysis suggesting type I respiratory failure. Mechanical ventilation was used to help the patient breathe. The serum sodium measurement was 103 mmol/l. Serological examinations determined that she was positive for M. pneumoniae (immunoglobulin (Ig)M antibody single titre 1:160). The cerebrospinal fluid (CSF) examination revealed normal intracranial pressure, cell count, protein, chloride and glucose levels. The CSF tests for infections caused by herpes simplex virus, cytomegalovirus, encephalitis B virus and varicella-zoster virus for M. pneumoniae were not performed. The serum and CSF were tested for AQP4-IgG using cell-based assays, demonstrating positivity in both assays. Autoimmune encephalitis antibodies (NMDAR, CASPR2, AMPAR, GABABR and LGI1) were examined, and the antibodies were all negative in the CSF and serum. The MOG-IgG, CSF OCB and IgG indicators were tested, producing negative results. Because the patient was experiencing muscle weakness, electromyography was performed to eliminate the presence of peripheral neuropathy. The electromyography result was normal. A pattern of increasingly slow waves was indicated in the electroencephalogram, and brain MRI revealed enhancing T2-weighted (T2W) hyperintense lesions in the hypothalamus and brainstem as well as meningeal enhancement (Fig. 2) but the disappearance of hyperintensity lesions in the SCC. Spinal MRI revealed an extensive T2 hyperintense signal from the C1–T7 and T12–L1 vertebral level (Fig. 3). The results of routine laboratory tests, including complete blood count, blood sugar, liver and renal function tests, coagulative function and electrocardiography, were all within normal limits. Other relevant routinely measured tests included rheumatic autoantibodies, antinuclear antibody, human immunodeficiency virus, syphilis, thyroid hormones, antiviral autoantibodies and tumour and paraneoplastic markers; all were negative. The tuberculin test was also negative.
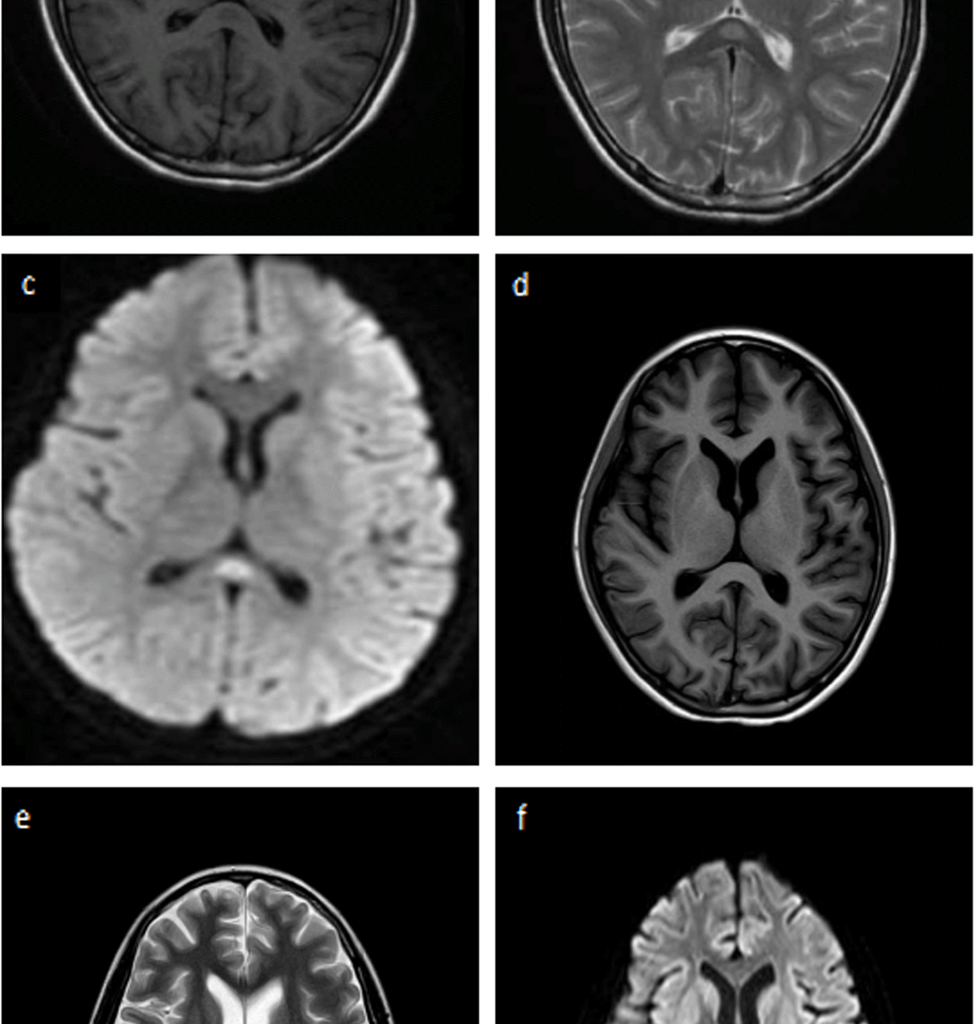
Fig. 1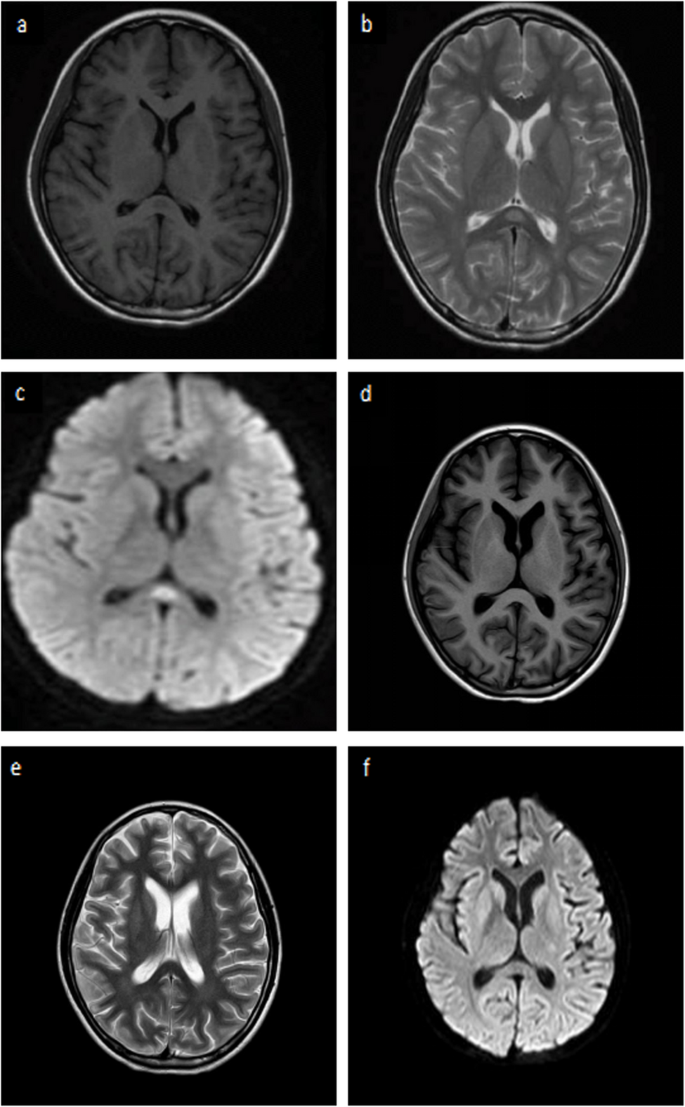
The brain magnetic resonance imaging (MRI). The brain MRI before admission revealed high intensity lesions (arrows) in the splenium of the collupus callosum (SCC) (a b c), which disappeared on one month (d e f)
Fig. 2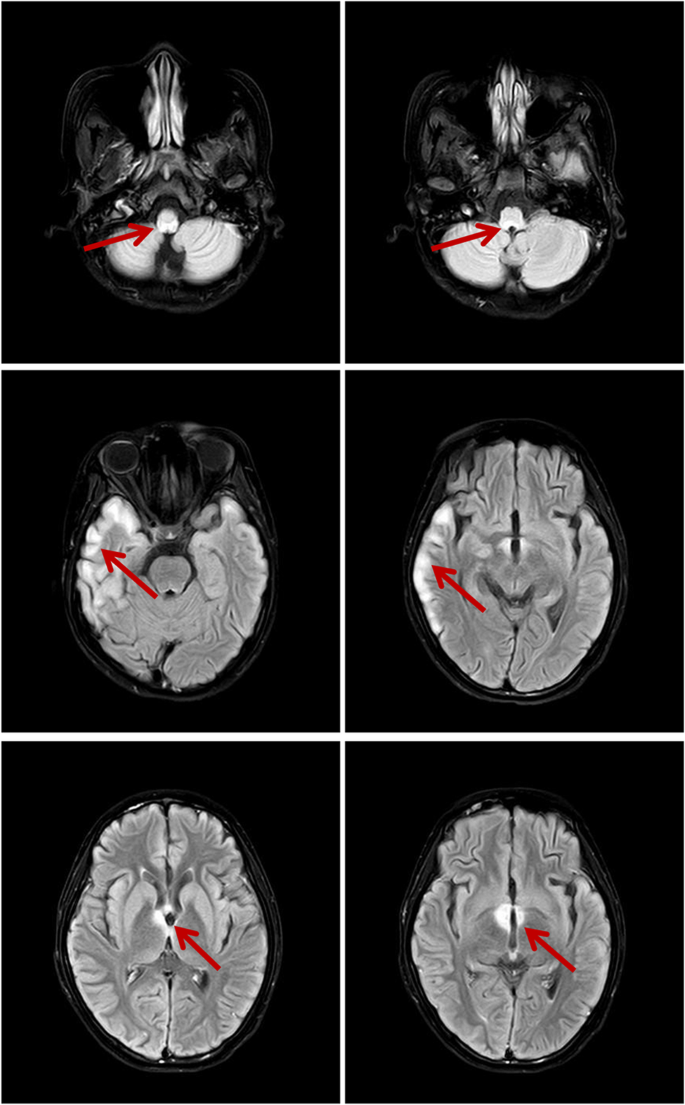
The brain MRI shows hyperintense lesion in hypothalamus, brainstem and meningeal enhancement
Fig. 3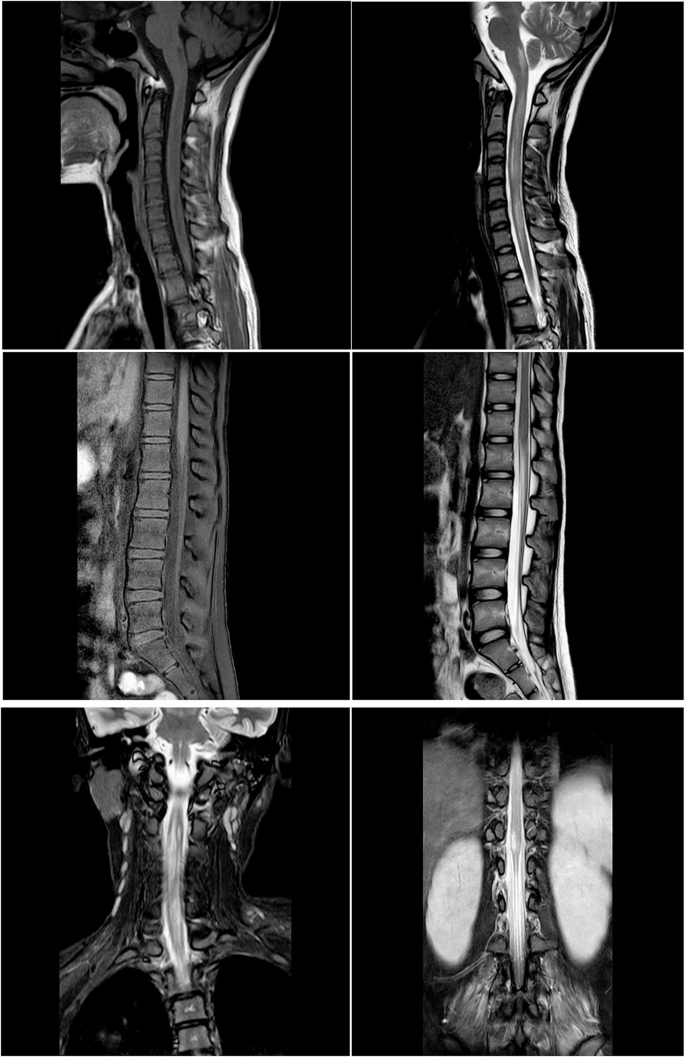
Spinal MRI showed the an extensive T2 hyperintense signal from C1 to T7、T12 to L1 vertebrae
After methylprednisolone therapy, the patient was removed from mechanical ventilation. She hiccupped after eating and had lower limb weakness that rapidly spread to the upper limbs. The upper and lower limb weakness was graded as Medical Research Council grade 1 and accompanied by paraesthesia. In addition, she had severe bladder dysfunction requiring catheterisation, but her visual function was normal. Thus, based on the clinical features, laboratory test results and MRI findings, a diagnosis of NMOSD with AQP4-IgG was confirmed.
Following diagnosis, methylprednisolone treatment was initiated. The starting dose was 800 mg/day for 3 days, which was then reduced to 600 mg/day for 3 days. For financial reasons, the patient refused the IVIG treatment. At the time of discharge, the patient’s urinary dysfunction had slightly improved. She did not fully recover her muscle strength, but the muscle strength of the left upper limb gradually returned to grade 3 and that of the right upper limb to grade 4 levels. The muscle strength of the lower limb gradually returned to grade 2 levels. The patient was discharged but continued to be treated with oral methylprednisolone. Due to the insufficient response to methylprednisolone therapy, immunosuppressive treatment with cyclophosphamide was initiated at a dose of 600 mg every two weeks for 5 months. It was changed to 600 mg/dose once/month at month 6 for a total duration of 12 months. Ophthalmic physical examination showed that visual acuity was 20/200 in the right eye and 20/40 in the left eye. Color vision was normal in the left eye and patient could not read Ishihara Color plates in the affected eye. Intraocular pressure measured 17 mmHg in the right eye and 14.1 mmHg in the left eye. Visual field testing revealed an inferior defect in the right eye and an inferotemporal defect in the left eye. At the final follow-up (3 months after onset), AQP-4 antibody was not detected after stabilisation of the patient and the patient did not experience further relapses, remaining clinically stable.
Discussion and conclusions
Our patient met the diagnostic criteria for AQP4-IgG-positive NMOSD based on the presence of area postrema syndrome, acute myelitis and compatible MRI findings, including longitudinally extensive transverse myelitis lesions (LETM) and brainstem lesions [2]. Notably, she initially presented with isolated lesions in the SCC and then the third ventricle. This finding is characteristic of reversible splenial lesion syndrome (RESLES), which may be associated with M. pneumoniae [3]. Although various infections have been reported to potentially trigger NMOSD [4], coexistence with M. pneumoniae is rare. Moreover, this patient developed hyponatraemia, which may be related to hypothalamic involvement. Previous studies have suggested that hyponatraemia is a potentially serious electrolyte abnormality observed in NMOSD, and its most common cause is syndrome of inappropriate antidiuretic hormone (SIADH) secretion [5, 6].
The pathogenesis of neurological complications in M. pneumoniae infection is incompletely understood but may involve direct invasion, autoimmune mechanisms or vasculopathy [7]. In this case, brain MRI revealed enhancing T2W hyperintense lesions in the hypothalamus and brainstem, as well as meningeal enhancement. This MRI presentation was considered to indicate meningeal involvement and could not exclude mycoplasma encephalitis. However, the diagnosis of Mycoplasma pneumoniae infection in our patient was based on a single elevated IgM antibody titer of 1:160. While serological testing remains one of the most widely used methods for diagnosing M. pneumoniae, especially in settings where PCR or CSF testing is not available, its diagnostic value can vary. Studies have shown that a single IgM titer may lack specificity, as false positives can occur due to cross-reactivity with other pathogens or persistent IgM following prior infection. The absence of M. pneumoniae screening in CSF is a limitation of this report. Tests for common pathogens (e.g. herpesvirus, bovine spongiform encephalopathy virus and cytomegalovirus) were performed to eliminate infection by these pathogens. Mycoplasma pneumoniae may have an indirect, likely autoimmune, mechanism rather than be the result of direct CNS invasion.
Notably, human AQP4 shares homologous epitopes with proteins from mycoplasma species, particularly in specific amino acid sequences and conformational structures [8]. These shared epitopes, especially within the extracellular loop domains of AQP4, may induce the immune system to produce antibodies against Mycoplasma that cross-react with human AQP4. This cross-reactivity can lead to disruption of the blood-brain barrier and trigger central nervous system inflammation, ultimately contributing to the development of NMOSD [9]. The molecular mimicry research shows that the second extracellular loop (ECL2) region of AQP4 is a key immunogenic target exhibiting structural similarity to corresponding sequences in Mycoplasma proteins, which facilitates this autoimmune cross-reaction [10]. Other autoantibodies have been found in the sera and CSF of patients with NMOSD, including antinuclear antibodies, SS antibodies and, in particular, antimyelin oligodendrocyte glycoprotein (MOG) antibodies [11]. Studies have also reported NMOSD following pulmonary tuberculosis in patients in Cape Town, South Africa, or mycoplasma pulmonary infections [12]. Aquaporins expressed by Mycobacterium and Mycoplasma sp. have been identified with similar residues to human AQP4 (unpublished observations, Wade WF) [11]. Additionally, pro-inflammatory cytokines such as IL-6, which are elevated in mycoplasma-associated RESLES [13], have been implicated in the pathogenesis of NMOSD. Therefore, M. pneumoniae infection may act as a trigger for AQP4-antibody-positive NMOSD through multiple immune-mediated mechanisms.
Our case, in line with the limited number of reports [14], suggests that M. pneumoniae infection may represent an underrecognised cause or trigger of NMOSD. Clinicians should consider this possibility in patients presenting with NMOSD, especially in the paediatric population. Further studies are warranted to clarify the strength of this association and the underlying pathogenic mechanisms. A better understanding of the link between infections and NMOSD may provide insights into disease pathogenesis and guide prevention strategies.