
Conjugated molecules, essential components in emerging technologies like organic electronics and solar energy, present a significant computational challenge for scientists, demanding efficient yet accurate modelling techniques. Marcel D. Fabian, Nina Glaser, and Gemma C. Solomon, all from the NNF Quantum Computing Programme at the Niels Bohr Institute, University of Copenhagen, address this need by revisiting the established Pariser-Parr-Pople (PPP) model, a simplified approach to understanding the behaviour of electrons within these complex systems. Their work demonstrates how this minimal model, despite its inherent approximations, continues to provide valuable insights and enables calculations that would otherwise be impossible, particularly when considering the crucial role of electron correlation. By showcasing recent applications in high-throughput screening for materials designed for singlet fission and inverted energy gap molecules, the team highlights the PPP model’s ongoing relevance and potential for accelerating the discovery of advanced materials.
Polyenes, Conjugation, and Electronic Structure Theory
This extensive collection of references reveals a vibrant research landscape spanning theoretical and computational chemistry, materials science, and increasingly, quantum computing. The core of this work focuses on molecular electronic structure theory, with a significant emphasis on understanding conjugated molecules, particularly polyenes, and their excited state properties. A major theme throughout is the investigation of molecules and materials exhibiting inverted singlet-triplet energy gaps, a crucial area for organic electronics and OLEDs, with implications for efficient light emission through thermally activated delayed fluorescence. Researchers explore the dynamics of excited states, examining singlet and triplet states, intersystem crossing, and the factors influencing fluorescence and phosphorescence.
This theoretical work directly informs the design and understanding of materials for organic light-emitting diodes, solar cells, and other electronic devices. The increasing number of references to quantum computing signals a growing recognition of its potential to revolutionize the field, offering new avenues for solving complex chemical problems. This body of work demonstrates a clear progression from fundamental theoretical development to practical applications in materials science and organic electronics. The research is highly interdisciplinary, bridging chemistry, physics, materials science, and computer science. The growing emphasis on quantum computing suggests that this field is poised for even more exciting breakthroughs in the years to come.
PPP Hamiltonian, From Hückel to Approximations
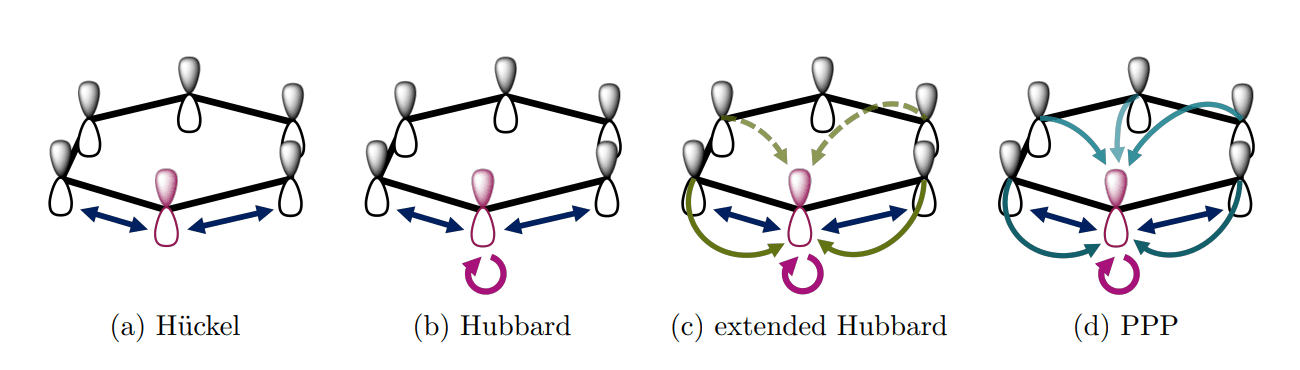
This study meticulously investigates the Pariser-Parr-Pople (PPP) Hamiltonian, a method for modelling conjugated electron systems, tracing its development from the foundational Hückel model. Researchers began by revisiting the core principles of the Hückel model, which simplifies calculations for conjugated molecules by distinguishing between σ- and π-molecular orbitals. This initial approach provided qualitative understanding of π-electron systems with minimal computational effort. To overcome computational hurdles, scientists implemented approximations to manage the complexity of electron-electron interactions.
Rather than explicitly calculating these interactions, the study focused on semi-empirical parameters and the zero differential overlap (ZDO) approximation, reducing the computational burden. Researchers systematically explored how these approximations impact the validity of the model, acknowledging the trade-offs between accuracy and computational feasibility. The work details a progression from qualitative understanding to increasingly sophisticated calculations, enabled by these methodological innovations. Scientists employed the PPP Hamiltonian to explore the electronic structure of conjugated systems, focusing on the behaviour of π-electrons within molecular frameworks. This approach allows for the investigation of systems that were previously intractable due to computational limitations.
PPP Hamiltonian Accurately Models Conjugated Systems
This research demonstrates the enduring power of the Pariser-Parr-Pople (PPP) Hamiltonian as a foundational model for understanding conjugated systems, revealing its continued relevance in modern computational chemistry. Researchers rigorously compared the PPP model with other Hamiltonian approaches, including the Hückel and Hubbard models, illustrating how the PPP Hamiltonian represents a minimal viable parametrization for describing chemically relevant systems. The study confirms that the PPP model’s initial approximations, developed in 1953, have been theoretically validated by subsequent accurate ab initio calculations, demonstrating its surprising robustness and predictive power. Researchers established that the PPP model effectively reduces the computational complexity of describing electron interactions, simplifying calculations from an impractical O(N 4 ) complexity to a manageable O(N 2 ) complexity, where N represents the system size. These advancements allow the model to treat three-dimensional systems with σ-electrons explicitly, broadening its applicability to a wider range of chemical structures.
PPP Model Validated for Conjugated Systems
This research demonstrates the continued utility of the Pariser-Parr-Pople (PPP) Hamiltonian as a model for conjugated π-electron systems, offering a computationally efficient approach to understanding their electronic properties. The work highlights how, despite being a semi-empirical method, the PPP model frequently achieves a level of agreement with experimental data comparable to, and sometimes exceeding, more computationally demanding ab initio calculations. Detailed analysis validates the core approximations within the PPP model, including the focus on π-electrons and the use of simplified overlap integrals, through comparison with a formally exact effective valence shell Hamiltonian. The research confirms that these approximations, while introduced ad hoc, are supported by a more rigorous theoretical framework. The authors acknowledge the inherent limitations of a semi-empirical approach, but demonstrate its continued relevance, particularly in high-throughput screening and inverse design problems related to materials science, specifically in the areas of singlet fission and singlet-triplet energy gap molecules.
👉 More information
🗞 The PPP model – a minimal viable parametrisation of conjugated chemistry for modern computing applications
🧠 ArXiv: https://arxiv.org/abs/2510.04632