
We identified 98 individuals with pathogenic or likely pathogenic GRIN2B variants from 23 publications [11, 16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37]. Additionally, we reported the clinical data for seven novel cases harboring likely pathogenic or pathogenic GRIN2B variants.
Patients with GRIN2B variants from our center
We recruited seven new cases from our center, four of which were female. These seven cases carried five novel variants: p.Phe554Ser, p.Ile641Thr, p.Asn649Ser, p.Val821Phe, and p.Gly1182Arg as well as two previously published variants: p.Met818Leu and p.Gly820Val [11, 22]. All seven variants were de novo and were classified as likely pathogenic or pathogenic according to the ACMG guidance. Four variants were located in the M4 transmembrane helix, two in the M3 transmembrane helix, and one in the M1 transmembrane helix. Of the 7 cases, 4 (57.14%) patients presented with epilepsy. Of these patients, only one patient who carried the p.Ile641Thr variant achieved seizure freedom after receiving memantine. All seven cases exhibited severe DD/ID and hypotonia. Microcephaly was observed in two cases (28.57%). Brain MRI results were available for six cases, five of which (83.33%) had abnormal results. Table 1 summarizes this data.
Table 1 Genetic and clinical characteristics of cases carrying GRIN2B variants from our hospitalClinical characteristics of individuals with reported and novel GRIN2B likely pathogenic or pathogenic variants
After combining our novel variants and those reported in the literature, we found 105 cases with GRIN2B variants. Information about gender was available for 76 cases, 40 (52.63%) of which were males. The mean age at inclusion and publication was 5.7 (range, 0–30) years. Of the 81 patients with available epilepsy-related information, 53.08% (43/81) presented with seizures. The mean age at seizure onset was 22.69 (range 0–108) months. The types of seizures included generalized seizures (13/36, 36.11%), focal seizures (15/36, 41.67%), infantile spasms (9/36, 25%), and epileptic spasms (9/36, 25%). Information about seizure outcomes was available for 27 patients. Of those patients, 25.93% (7/27) achieved seizure control. Abnormal EEG results were found in 87.93% (51/58) of cases. Of the 105 cases, all (100.00%) presented with DD/ID. Of those, 71 (67.62%) cases had known severity. Among the individuals with known DD/ID severity, mild manifestations were observed in 15.49% (11/71) of the cases, moderate manifestations in 19.72% (14/71), severe manifestations in 63.38% (45/71). Of the 81 patients for whom information about ASD was available, 25 (30.86%) patients had it. Other clinical manifestations included movement and motor disorders (43/105, 40.95%), spasticity (6/41, 14.63%), hypotonia (48/55, 87.27%), microcephaly (10/55, 18.18%), and language impairment (48/105, 45.71%). Among individuals with language impairment, 64.58% (31/48) could not speak. Brain MRI results were available for 62 cases, 25 of which had abnormal results, including widened ventricles (n = 6), polymicrogyria (n = 5), delayed myelination (n = 4), and hypoplastic corpus callosum (n = 4). Other anomalies included an abnormal cerebellum (n = 2), cortical atrophy or dysplasia (n = 4), basal ganglia dysplasia (n = 3), an abnormal signal in the left amygdala (n = 1), a widened subarachnoid space in the bilateral frontotemporal regions (n = 1), and increased extra-axial spaces (n = 1). Supplementary Table 1 summarizes these results.
A total of 84 likely pathogenic or pathogenic variants were identified in the 105 cases. Of the 84 variants identified, ten were recurrent: p.Gly820Ala (n = 6), p.Gly689Ser (n = 6), p.Arg847* (n = 4), p.Ile751Thr (n = 3), and p.Met818Leu (n = 2), p.Ala734Val (n = 2), p.Arg540His (n = 2), p.Arg696His (n = 2), p.Gly820Val (n = 2), and p.Lys1091Thr (n = 2). There were 69 missense variants and 16 truncated variants (Supplementary Table 1). The p.Met818Leu variant was observed in two males presenting with early onset epileptic spasms, severe DD, hypotonia, and visual impairment. One of the patients was seen at our center. He was treated with levetiracetam (LEV), phenobarbital (PB), topiramate (TPM), L-serine, and perampanel; however, he continued to have refractory epilepsy. The second patient was found in the literature and was treated with LEV, clobazam (CLB), TPM, and prednisolone, but did not improve. However, memantine reduced seizure frequency by 80%. The p.Gly820Ala variant was found in six patients presenting with heterogeneous phenotypes: DD/ID (n = 6), ASD (n = 2), movement disorders (n = 2), and refractory infantile spasms (n = 1). The patient with infantile spasms was treated with valproic acid (VPA), which did not improve the condition. Although carbamazepine (CBZ) led to a temporary improvement, the patient remained with uncontrolled seizures. The p.Arg847* variant was identified in four individuals with various manifestations: DD/ID (n = 4), ASD (n = 1), and hypotonia (n = 2). The p.Gly689Ser variant was found in six individuals presenting with DD/ID (n = 6), hypotonia (n = 4), movement disorders (n = 4), and seizures (n = 3). One patient with a seizure disorder received treatment with vigabatrin (VGB), which increased seizure frequency. Treatment with TPM led to a partial improvement, while the patient still experiences ongoing seizures. The p.Ile751Thr variant was found in three cases who presented with DD/ID (n = 3), ASD (n = 1), and hypotonia (n = 3).
Clinical features of patients with LOF variants and GOF variants, and clinical indicators for LOF and GOF variants
Electrophysiological channel characterization was conducted for 41 out of 85 (48.24%) of all reported GRIN2B variants. Previous studies have described 42 different LOF electrophysiological variants, including 18 in the ABD domain, seven in the TMD, and three in the CTD [11, 21, 34, 36, 38,39,40,41]. Previous studies [11, 39], found that thirteen different variants caused a GOF effect in cellular expression systems, including five variants in the ABD domain and eight in the TMD domain. Furthermore, variants in the TMD domain were more likely to be GOF (P < 0.05). As previously proposed, we grouped patients according to the known functional consequences on NMDARs for phenotypic comparison (Table 2, Supplementary Table 2).
To date, 42 different GRIN2B LOF variants have been identified in 55 cases, and 14 different GOF variants have been identified in 16 cases. Of the 55 patients with LOF variants, 48 had information about epilepsy. Of those, 41.67% (20/48) experienced seizures. The mean age at seizure onset was 12 months (range: 0–108). Types of seizures included generalized (5/14, 35.71%), focal (3/14, 21.43%) seizures, both focal and generalized (2/14, 14.29%), myoclonic seizures (4/14, 28.57%), and infantile spasms (1/14, 7.14%). Information about seizure outcomes was available for 17 patients. Of those patients, 29.41% (5) achieved seizure control. Abnormal EEG results were found in 82.35% (28/34) of cases. All 55 cases (100.00%) presented with DD/ID. Of the 38 cases with known DD/ID severity, mild manifestations were observed in seven (18.42%) patients, moderate in eight (21.05%) patients, and severe in 23 patients (60.53%). ASD was observed in 29.00% (16/55) of patients. Additional clinical manifestations included movement and motor disorders (45.45%, 25/55), language impairment (49.09%, 27/55), and microcephaly (14.29%, 4/28). Of the patients with language impairment, 59.26% (16/27) could not speak. Brain MRI results were available for 35 cases, 13 of which had abnormal results, including widened ventricles, delayed myelination, cortical atrophy, and hypoplastic corpus callosum. In addition, LOF missense variants were found to be associated with more severe clinical phenotypes than LOF truncated variants. These severe clinical manifestations included severe DD/ID, speech impairment, and movement disorders (Table 3, Supplementary Table 5).
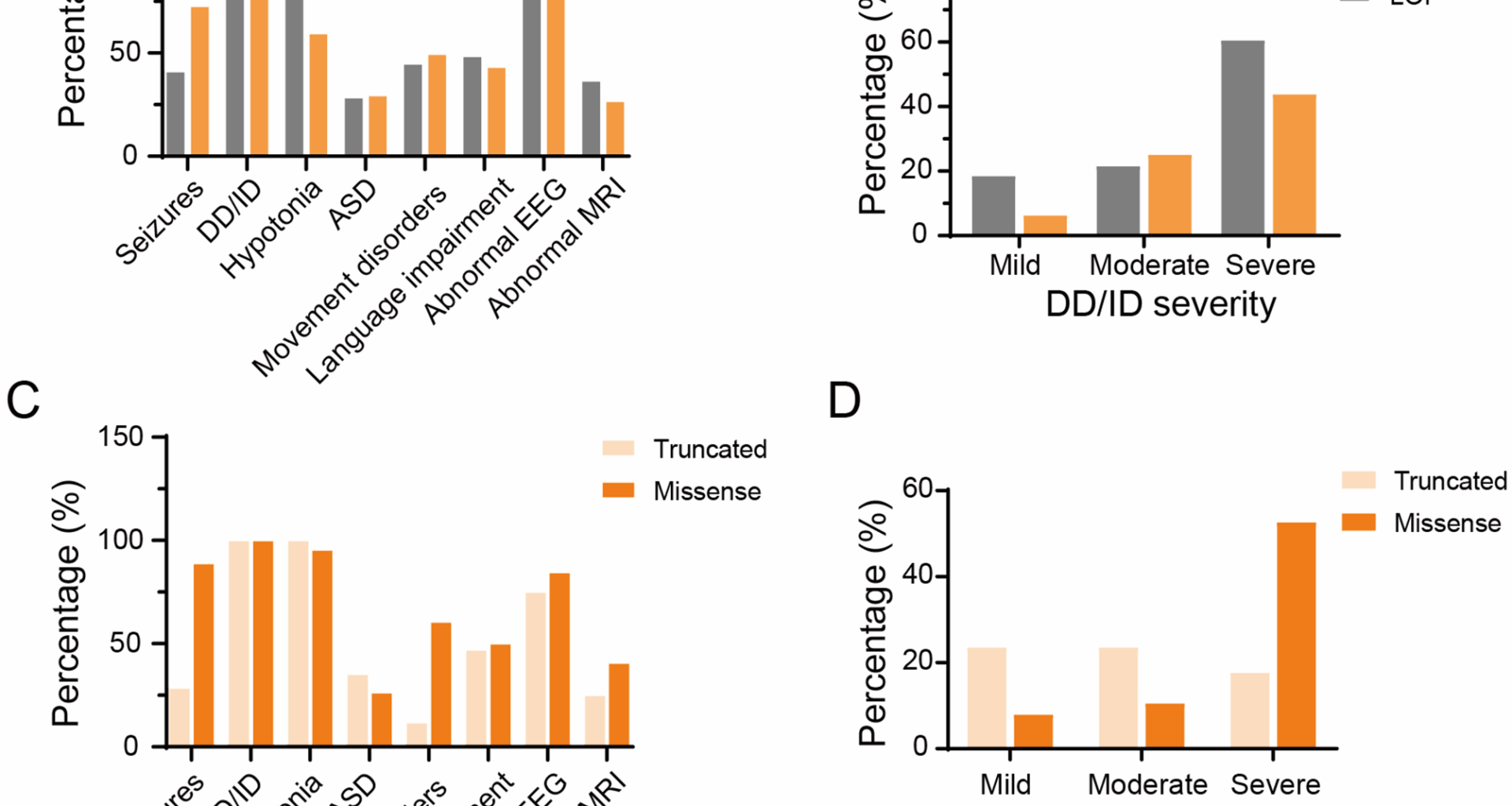
Of the 16 patients with GOF variants, 15 had information about epilepsy, 11 of whom (73.33%) presented with seizures. The mean age at seizure onset was 3.5 (range, 0-117) months. The types of seizures included generalized seizures (2/10, 20%) and focal seizures (1/10, 10%). Of the eleven patients with information about seizure outcomes, only one achieved seizure control. The EEG was abnormal in 90.91% (10/11) of cases. All 16 cases (100%) presented with DD/ID. Of the 12 cases with known DD/ID severity, one patient had a mild form, four patients had a moderate form, and seven patients had a severe form. Five out of 15 patients (33.33%) presented with ASD. Additional clinical manifestations included movement and motor disorders (8/16, 50%), language impairment (7/16, 43.75%), and microcephaly (5/16, 31.25%). Of the patients with language impairment, 85.71% (6/7) had no speech. Brain MRI results were available for 11 cases, 3 of which (27.27%) had abnormal results, including severe cortical and central atrophy, delayed myelination, and polymicrogyria of the optic nerves and optic chiasm. Figure 1 summarizes these results.
Fig. 1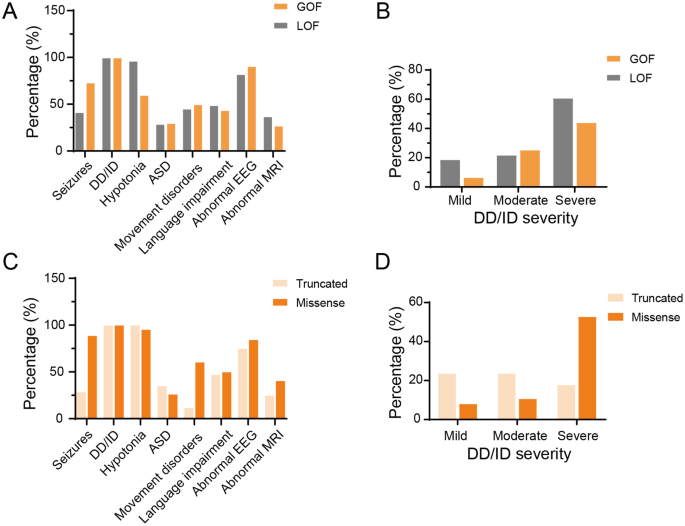
Distinctive phenotypic features of GRIN2B-related neurodevelopmental disorders in individuals with different type of variants. (A, B) Representation of the main phenotypic features in patients carrying GOF versus LOF variants. Values are presented as the percentage of patients exhibiting each trait, calculated relative to the number of patients with available data. (C, D) Representation of the main phenotypic features in patients carrying LOF missense versus truncated variants. Abbreviations: DD: developmental delay, ID: intellectual disability, EEG: electroencephalograph, ASD: autism spectrum disorder, MRI: magnetic resonance imaging
Compared to individuals with LOF variants, patients with GOF variants were more likely to experience seizures of various severities and microcephaly (p < 0.05). However, there was no significant difference in seizure onset, seizure frequency, or epilepsy outcome among the groups. Furthermore, no substantial disparities were observed between the two groups in other phenotypes, encompassing DD/ID, language impairment, movement disorder, ASD, and brain MRI and EEG abnormalities (Table 2).
Table 2 Comparison of clinical features between patients with GOF variants and those with LOF variantsTable 3 Comparison of clinical features between the patients with LOF missense variants vs patients with LOF protein truncating variantsTreatment of GRIN2B-related epilepsy: conventional anti-seizure medications and memantine
Treatment information was available for 26 cases, 21 of which were from the previous study and five of which were from this study (Supplementary Tables 3, 4). Of the 26 patients, 10 patients (38.46%) were refractory to ASMs, five patients (19.23%) had a partial response, and 11 patients (42.31%) achieved seizure freedom. The seizure freedom proportions for effective drug combinations were as follows: ACTH/prednisolone (3/3); lamotrigine (1/1); PB (1/1); TPM (1/4); VPA (0/4); oxcarbazepine (0/2); CBZ (0/1); sulthiame (0/1); CLB (0/1); VGB (0/1); midazolam (0/1); and LEV (0/1). The efficacies of the therapies used for epilepsy, either alone or in combination, were as follows: ACTH/prednisolone (60%, 3/5); TPM (30%, 3/10); LTG (40%, 2/5); VPA (8.33%, 1/12); CBZ (25%, 1/4); VGB (14.29%, 1/7); PB (33.33%, 1/3); and L-serine (50%, 1/2). Furthermore, rufinamide and the ketogenic diet were reported to be effective in one case each. However, LEV, perampanel, midazolam, CLB, and OXC were ineffective (Fig. 3). Of the nine patients who presented with infantile spasms, three were administered ACTH. Two of these patients (66.67%) achieved partial seizure control, while one patient (33.33%) showed a poor response, likely due to the presence of status epilepticus. We administered ACTH to our two patients diagnosed with infantile spasms. However, they experienced only a transient reduction in seizures, with no cognitive or developmental improvement.
Besides, memantine effectively controlled seizures and improved cognition in patients with GRIN2B-related developmental disorders. Two patients in our cohort received memantine treatment, which resulted in seizure control and EEG normalization (Fig. 2). In addition, we observed cognitive improvement in some domains, including attention, eye contact, and social interaction in our two patients. In patient 2, a two-month memantine regimen elicited marked improvements in social reciprocity, as evidenced by enhanced social smiling and sustained attention. For patient 4, memantine administration resulted in partial responsiveness to auditory stimuli and slight gains in attention. None of our patients experienced adverse drug effects. Of the seven patients (including those from the literature) who were treated with memantine, six experienced cognitive improvement (85.71%), and three achieved seizure control (42.86%) (Fig. 3 summarizes this information).
Fig. 2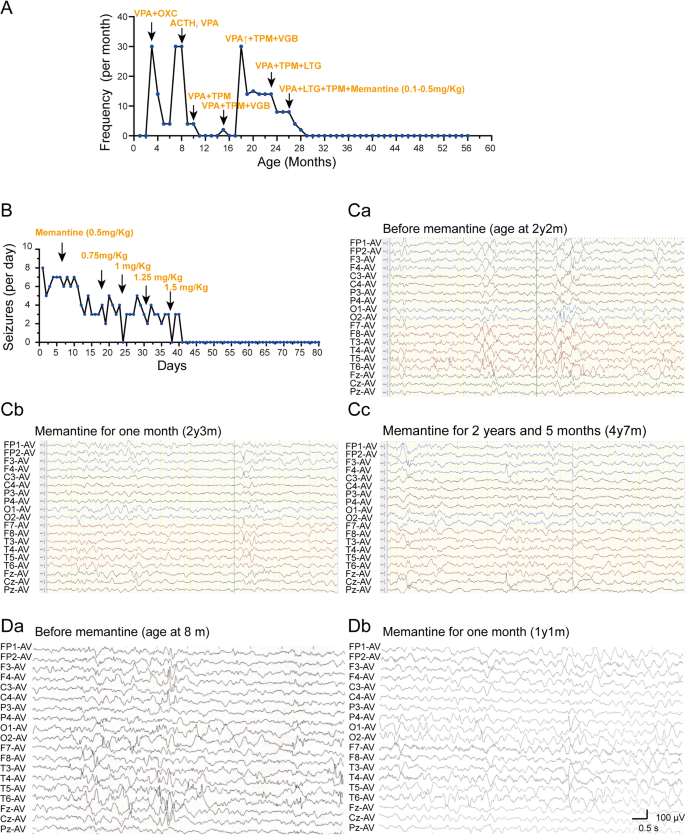
The efficacy of anti-seizure medications and memantine was assessed in two patients harboring distinct variants in the TMD region. (A, B) Seizure counts for the patients with the p.Ile641Thr and p.Met818Leu variants, respectively. (Ca-Cc) Electroencephalographic (EEG) recordings of patients carrying the p.Ile641Thr variant before (Ca) and after the application of memantine (Cb, Cc). All three EEG recordings show multifocal spike waves, spike-and-sharp waves, slow-spike waves, and sharp waves, prominently in the posterior regions. Post memantine EEGs (Cb, Cc) demonstrate a significant reduction in spike waves compared with Ca. Notably, epileptic spasms were observed in the pre-treatment EEG. (Da-b) EEG recordings of patients carrying the p.Met818Leu variant before (Da) and after (Db) the application of memantine. Da, EEG recordings show multifocal spike waves, sharp waves, slow-spike waves, and sharp waves predominantly in occipital and temporal regions. Db, EEG of post memantine reveals a slight reduction in spike waves when compared to Da
Fig. 3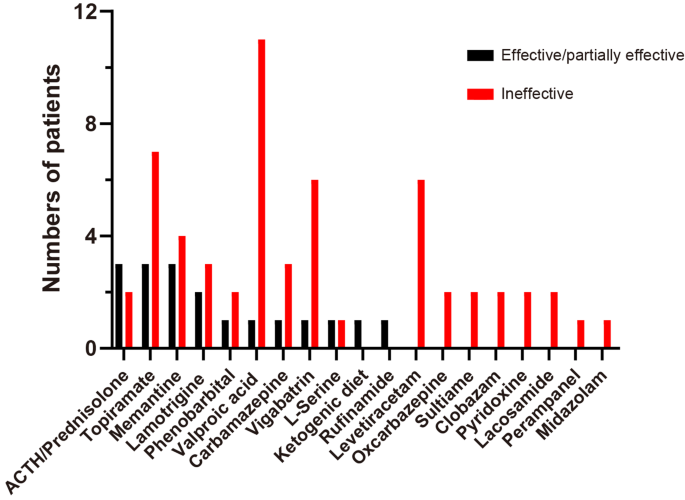
The bar graph summarizes the treatments used for epilepsy and their efficacy. Abbreviations: ACTH, adrenocorticotropic hormone