
Physicochemical characteristics of soil of five locations in Tunisia
As described in the Materials and Methods, we assessed the physicochemical properties of the soil at a depth of 0–15 cm in five plots in Tunisia (Table 1). ANOVA and Tukey’s test results showed significant differences between sites, particularly between S2 and the others (Additional file 4). The soils were predominantly sandy (S2) and sandy loam (other sites), generally very dry and alkaline. Electrical conductivity exceeded 2000 µS/cm, indicating high salinity at S2 and S4, and extremely high salinity at S1, S3, and S5. Nitrate levels were excessive across all sites, while organic matter was low at S2, medium at S1, S4, and S5, and high at S3. The C/N ratio, above 25% for most sites, suggests slow decomposition of organic matter, except at S2, where it was deficient. All sites were rich in CaCO3 and classified as calcareous soils. Available phosphorus was low at S2 and S3, and medium at S1, S4, and S5. Overall, S3 and S1 were similar for most parameters.
Table 1 Geographic coordinates and soil properties of sampling sites. Each value represents the mean ± standard deviation (SD) of five replicate soil profiles per siteMolecular identification of collected plants
It was of interest to determine the genetic diversity of the collected plants to provide accurate identification, uncover cryptic species, and assess the ecological and adaptive characteristics of the vegetation in the selected sites. Fragments of approximately 700 bp (Additional file 5) were amplified corresponding to the intergenic regions ITS1 and ITS4, and the gene encoding the 5.8 S rRNA. The sequences of the 22 accessions were submitted to NCBI GenBank (Accession numbers: OR473670-OR473692 for the entire ITS region) (Table 2). ITS1-4 revealed 9 different families: Zygophyllaceae, Nitrariaceae, Aizoaceae, Ephedraceae, Boraginaceae, Orobanchaceae, Amaranthaceae, Convolvulaceae, and Plumbaginaceae.
Table 2 Geographical origin and accession numbers of 22 collected plantsAmplicon sequencing of endophytic communities in green and root compartments of halotolerant plants
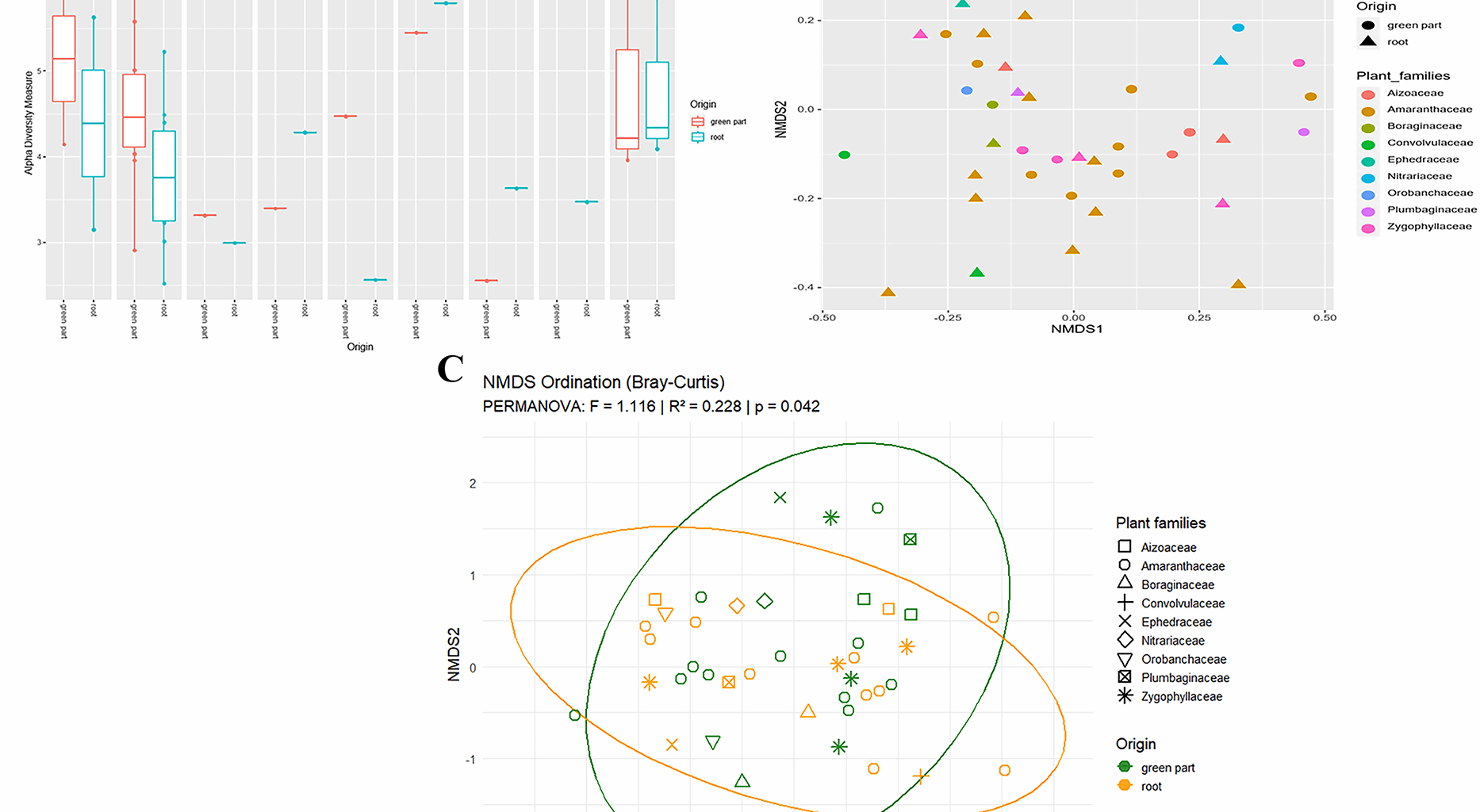
It was of interest to analyze the endophytic bacterial community inhabiting the collected plant samples. After filtering, the dataset comprises a total of 7,623,850 reads across all samples, with an average of approximately 103,025 reads per sample, ranging from 27,693 to 252,690. The total number of reads associated with mitochondria is 8699, with an average number of reads per sample of 119, ranging from 0 to 539 reads per sample. Therefore, the total number of reads associated with the chloroplast is 539,205, with an average number of reads per sample of 7386, ranging from 0 to 5738 reads per sample. The samples were rarefied to a sampling depth of 6000 reads per sample (Additional file 6). The total number of taxa was 10502, distributed in 33 phyla, 98 classes, 236 orders, 353 families, and 648 genera. The alpha diversity calculated using the Shannon diversity index for different plant families, and categorized by the plant compartment (i.e. green part, and root) ranged from 2.5 to 6 and showed moderate differences between the origin of the microbiome sample collection (Fig. 1A). The overall trends displayed that the green part consistently showed the highest Shannon diversity index across all plant families, indicating a higher microbial richness and evenness in this part of the plant compared to the root compartment. Significant differences in the alpha diversity were observed only between plants collected in site 1 and site 5 (p-value = 0.0500), especially in the green compartment (Additional file 7), suggesting that the location does not interfere strongly with the endophytic microbiome biodiversity of the halophytic plants. In line with the results of the alpha diversity, the beta diversity, calculated by using the Bray-Curtis dissimilarity measure, revealed that the green and the root samples were relatively similar (Fig. 1B), and no clear clustering was detected based on the plant families. However, PERMANOVA analysis (Fig. 1C) revealed a statistically significant effect of tissue origin (green part vs. root) on microbial community structure (p = 0.042), regardless of plant family. Interestingly, endophytic microbiome composition differs between above- and below-ground tissues within the same plant family. This suggests that the plant compartment plays a role in shaping the endophytic microbiome, which is nonetheless composed of bacterial members well adapted to the shared environmental conditions, regardless of plant family or soil origin. To assess the differences in microbiome composition at the phylum level between various plant families and between green parts and roots, a plot illustrating the relative abundance of the predominant taxonomic groups was generated (Fig. 2). Major bacterial populations in halophilic plants included Proteobacteria (67.80%), Firmicutes (14.06%), and Actinobacteria (6.57%). Proteobacteria were consistently dominant in both aerial green parts and roots, but its proportion was higher in the green parts. Firmicutes were present in both green parts and roots, but often in higher relative abundance in roots, and Actinobacteria generally show higher relative abundance in roots. Variability within the phyla composition was more pronounced in roots, suggesting a more diverse microbiome compared to the green parts. Regarding the differences between the plant families, we observed that some families, like Amaranthaceae and Orobanchaceae, displayed more balanced distributions among the phyla, while others like Ephedraceae and Zygophyllaceae are dominated by Proteobacteria.
Fig. 1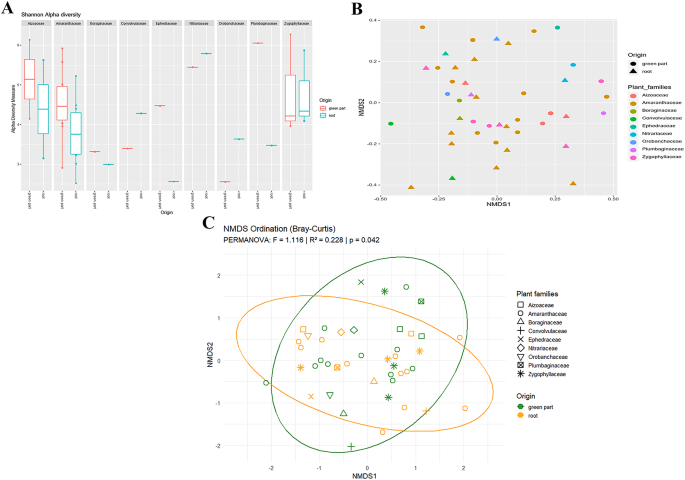
Diversity Metrics for Different Plant Families: A Non-Culturable Alpha Diversity Measured by Shannon Index. B Non-Culturable Beta Diversity Assessed by Bray-Curtis Dissimilarity. C NMDS Ordination Based on Bray-Curtis Dissimilarity Showing The Effect Of Origin and Plant Family on Non-Culturable Beta Diversity (PERMANOVA, p < 0.05)
Fig. 2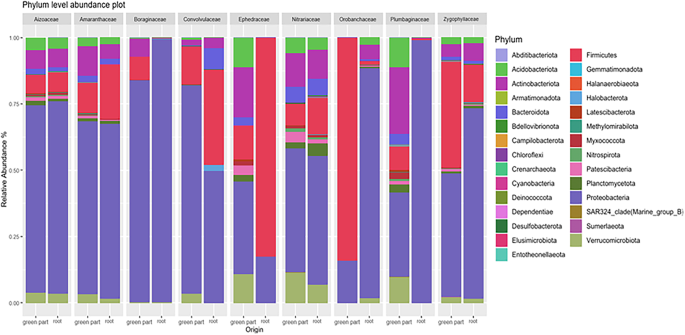
Relative Abundance of Predominant Taxonomic Groups Across Different Plant Families and Between Green Parts and Roots
A persistent group of endosphere-inhabiting taxa is consistently present across different halophytic plants
In order to define the members of the microbiome most adapted to the endosphere of halophytic plants, we empirically quantified the most prevalent taxa (MPT) or “common core” microbiome. At the genus level, we defined as MPT the genera that were detected in at least 60% of the samples and at > 0.5% relative abundance. A shortlist of the most prevalent genera was Acinetobacter, Halomonas, Kushneria, Pseudomonas, Psychrobacter, Stenotrophomonas, and Streptomyces (Fig. 3). All of these were present across all plant families, albeit with variations in their relative abundance. For example, the presence of Halomonas was notably higher in some plant families compared to others; Aizoaceae and Ephedraceae showed a significant presence of Halomonas, while Amaranthaceae and Boraginaceae presented a balanced community formed by various genera. Interestingly, root parts had an higher relative abundance of Halomonas and Pseudomonas compared to the green parts, indicating a possible preference or better adaptation of these genera for the root environment. On the other hand, green parts showed a higher presence of Psychrobacter and Stenotrophomonas in certain plant families, indicating possible niche preferences or specific plant-microbe interactions. Weissella, was detected exclusively in the root part of the Ephedraceae family and the green part of the Orobanchaceae family (data not shown). Although these plants belong to different sites, they shared this common feature. In summary, the barplot highlights the core microbiome composition across different plant families and plant compartments.
Fig. 3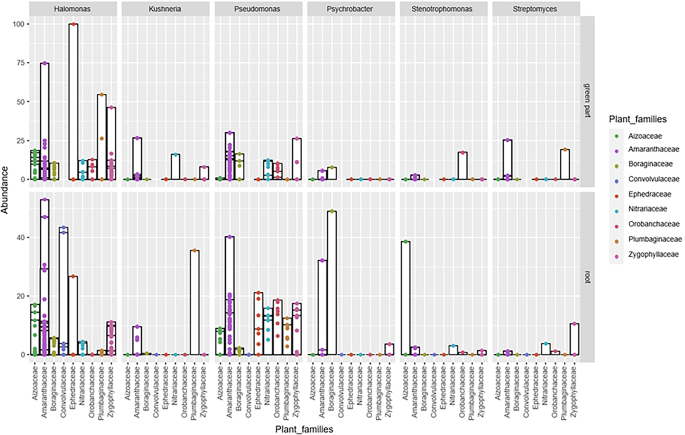
Most prevalent taxa (MPT) at the genus level across different plant families
The culturable fraction of endophytic bacteria across different halophytic plant varieties
The same targeted sequencing of 16 S rRNA gene was also applied to the cultured fraction of the microbiome to provide indicative information on the total number of potential culturable strains present across different halophytic plant varieties. With this aim, samples were grown under laboratory conditions and en masse collected to extract the total DNA to create the libraries. The results obtained showed that the bacterial communities of 22 collected plants (Fig. 4) were dominated by Proteobacteria (25 genera), followed by Firmicutes (13 genera), and Actinobacteria (11 genera). Halomonas, Pseudomonas, and Kushneria were the predominant genera among the Proteobacteria, while Bacillus, Terribacillus, and Oceanobacillus dominated the Firmicutes, with Kocuria being the main genus among Actinobacteria. Significant variations were observed in the relative abundance of Halomonas across different plants and sites, with a notable prevalence in plants from site 3. Oceanobacillus abundance also varied, being high in Halocnemum strobilaceum from site 4, but lower in other plants like Cistanche deserticola and Tetraena fontanesii. Bacillus genus showed variability too, with Suaeda fructicose from site 1 having a high abundance, while others like Suaeda nigra and Salicornia fruticose showed low values. Specific genera like Psychrobacter, Photobacterium, and Gracilibacillus were found exclusively in certain plants or in very low abundance in others.
Fig. 4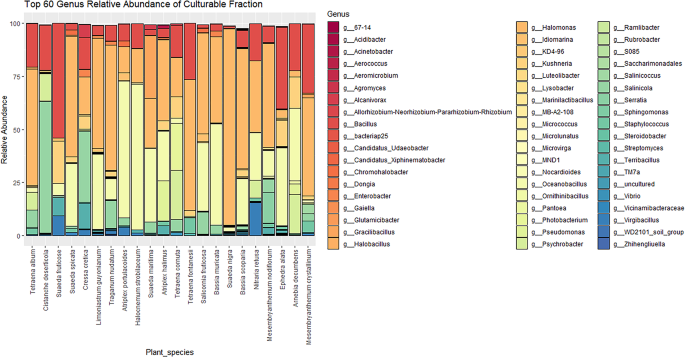
Relative Abundance of Culturable Endophytic Bacteria at the Genus Level Across Different Halophytic Plants
Culturable collection of halophytic endosphere-associated bacteria
A total of 902 culturable endophytic bacterial isolates were obtained from two plant compartments (aerial green part and roots) of 22 plants, as shown in (Fig. 5) and described in the Materials and Methods. The higher amounts of isolates were isolated from root compartments of Tetraena cornuta, Atriplex portulacoides, and Tetraena album at site 1, and Ephedra alata at site 2. Notably, Tetraena cornuta gave the highest number of isolates overall, with 82 isolates, 48 from roots and 34 from green parts. Atriplex portulacoides and Tetraena album gave 59 and 73 isolates, respectively. In contrast, most isolates from other plant species were predominantly obtained from the green parts, such as Limoniastrum guyonianum (39 isolates, mainly from green parts) and Mesembryanthemum crystallinum (59 isolates, with similar numbers from both compartments). The morphological and biochemical characterization of those culturable endophytic bacterial isolates is presented in (Additional file 8). The biochemical reactions differed among the isolates, while all of them exhibited positive catalase activity. These data allow for a preliminary classification of isolates and provide a global overview of their phenotypic diversity. They constitute an essential basis for more in-depth taxonomic or phylogenetic analyses.
Fig. 5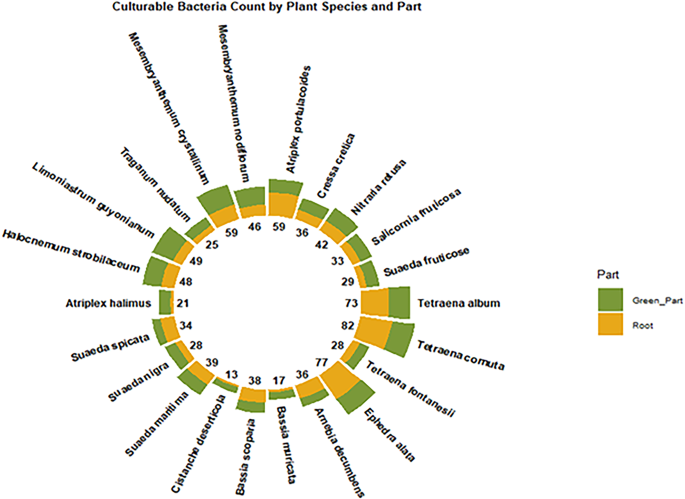
Distribution and Number of Culturable Endophytic Bacteria in Different Halophytic Plants
Canonical correspondence analysis (CCA) based on the top 60 genera and associated environmental factors
The canonical correspondence analysis (CCA) plot visually represents the relationships between species, soil chemical parameters, and sampling sites (Fig. 6). The first and second canonical axes explained 41.88% (eigenvalue of 0.28) and 27.69% (eigenvalue of 0.18) of the variation in the species data, respectively. Several bacterial genera aligned with the positive side of CCA2, where available phosphorus (AP) was also positioned, indicating a spatial correspondence between AP and phosphate-solubilizing bacteria (PSB) such as Terribacillus, Dongia, Streptomyces, Gaiella, Rhizobium, and Luteolibacter. The genera Halomonas, Achromobacter, Stenotrophomonas, Serratia, Salinicoccus, Brevundimonas, and Aliidiomarina are closely associated with site 5, which is mainly influenced by several soil chemical parameters, including EC (Electrical Conductivity), TC (Total Carbon), OM (Organic Matter), MC (Moisture Content), TCa (Total Calcium), and NO₃⁻ (Nitrate). This association suggests that these bacteria are adapted to environments with elevated salinity, nitrate, and organic content. Furthermore, the CCA graph pointed out that the communities present in site1 and site4 were related to pH and TN (Total Nitrogen). Among the genera influenced by these parameters, we can mention Pseudomonas, Aerococcus, Photobacterium, Kushneria, and Chromohalobacter. Although genera such as Bacillus, Paenibacillus, Alcanivorax, Salinicola, Psychrobacter, Enterobacter, and Erwinia were mainly associated with site2 and site3 in the CCA plot, they did not show any clear correspondence with the measured soil parameters. This may suggest a broad ecological tolerance, meaning they do not exhibit strong preferences for specific soil conditions.
Fig. 6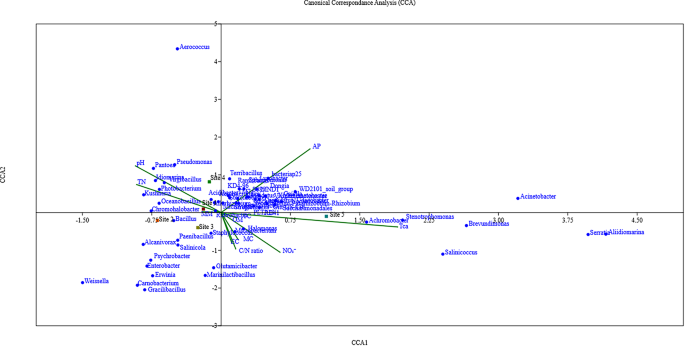
Canonical Correspondence Analysis (CCA) of Metagenomic Sequence Data and Environmental Factors. Green lines represent soil parameters. Only the top 60 genera are shown. Names in blue indicate bacterial genera, and colored squares represent sampling sites
Functional predictions of bacterial communities
This analysis employed PICRUSt to investigate the functional pathways of endophytes in extremophilic plants for tolerance to biotic and abiotic stresses. An enriched functional prediction was found, suggesting that the microbiomes inhabiting extremophilic plants were significant. A total of 425 pathways were identified from the 10,126 ASVs of the study, which were categorized into several functional modules. The most abundant included amino acid metabolism, lipid metabolism, energy metabolism, metabolism of cofactors and vitamins, nucleotide metabolism, and fatty acid biosynthesis. Among these, pyruvate fermentation to isobutanol and L-isoleucine biosynthesis II were the most frequent functions across all plant families. The heatmap showed strong clustering between Ephedraceae and Orobanchaceae due to their metabolic similarities, while Amaranthaceae and Convolvulaceae had higher metabolite abundance (Fig. 7A). The clustering of the Ephedraceae and Orobanchaceae families, due to their high functional metabolic similarities, was reflected in the seven highest detected pathways, which were exclusive to these two families. This unique presence may be attributed to the abundance of the Weissella genus. Specific functions, such as adenosine nucleotides degradation IV, the superpathway of CDP-glucose-derived O-antigen building blocks biosynthesis and 7-(3-amino-3-carboxypropyl)-wyosine biosynthesis, were exclusive to the Convolvulaceae family, whereas the Nitrariaceae family uniquely exhibited functions like the 3-hydroxypropanoate cycle, glyoxylate assimilation and the superpathway of the 3-hydroxypropanoate cycle were uniquely observed in the Nitrariaceae family. Additionally, chondroitin sulfate degradation I (bacterial) was found only in the Zygophyllaceae family. Unique functions were identified in specific families, suggesting that differences between plant families are driven more by endophytic bacterial communities than by site variability (Fig. 7B).
Fig. 7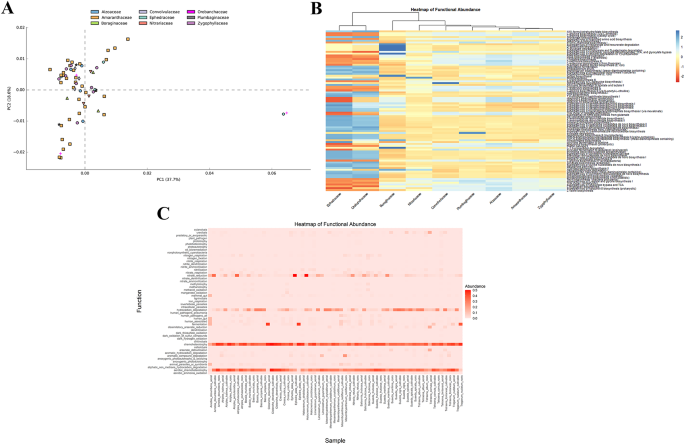
Functional Pathway Profiles of Endophytic Communities Across Plant Families: A PCA plot comparing functional pathway profiles of 9 plant families. B Heatmap of predicted functional pathways related to endophytic communities of extremophilic plants by PICRUSt2. C Relative abundances of FAPROTAX functional groups
Based on the FAPROTAX analysis, 7 biological metabolic pathways were detected, including nitrogen cycle functions, carbon cycle and carbon degradation, sulfur and other elemental cycles, photosynthesis functions, host-associated functions, hydrocarbon degradation (bioremediation), and polymer degradation (plant biomass), as well as 51 subfunctional groups (Fig. 7C). Among the lesser abundant predicted traits, a majority were assigned to nitrogen cycle functions and host-associated functions. Nitrogen cycle functions were the dominant category, with 13 functions, such as nitrate reduction, nitrogen fixation, nitrate respiration, nitrogen respiration, and nitrification. For host-associated functions, 10 functions were detected, namely intracellular parasites, plant pathogens, Human pathogens, and animal parasites. Aerobic chemoheterotrophy and chemoheterotrophy were the most dominant functional groups in all samples, followed by hydrocarbon degradation and nitrate reduction. Additionally, genes related to the fermentation function were remarkably abundant in the aerial part of Cistanche deserticola and the root part of Ephedra alata and Traganum nudatum compared with the other samples.