
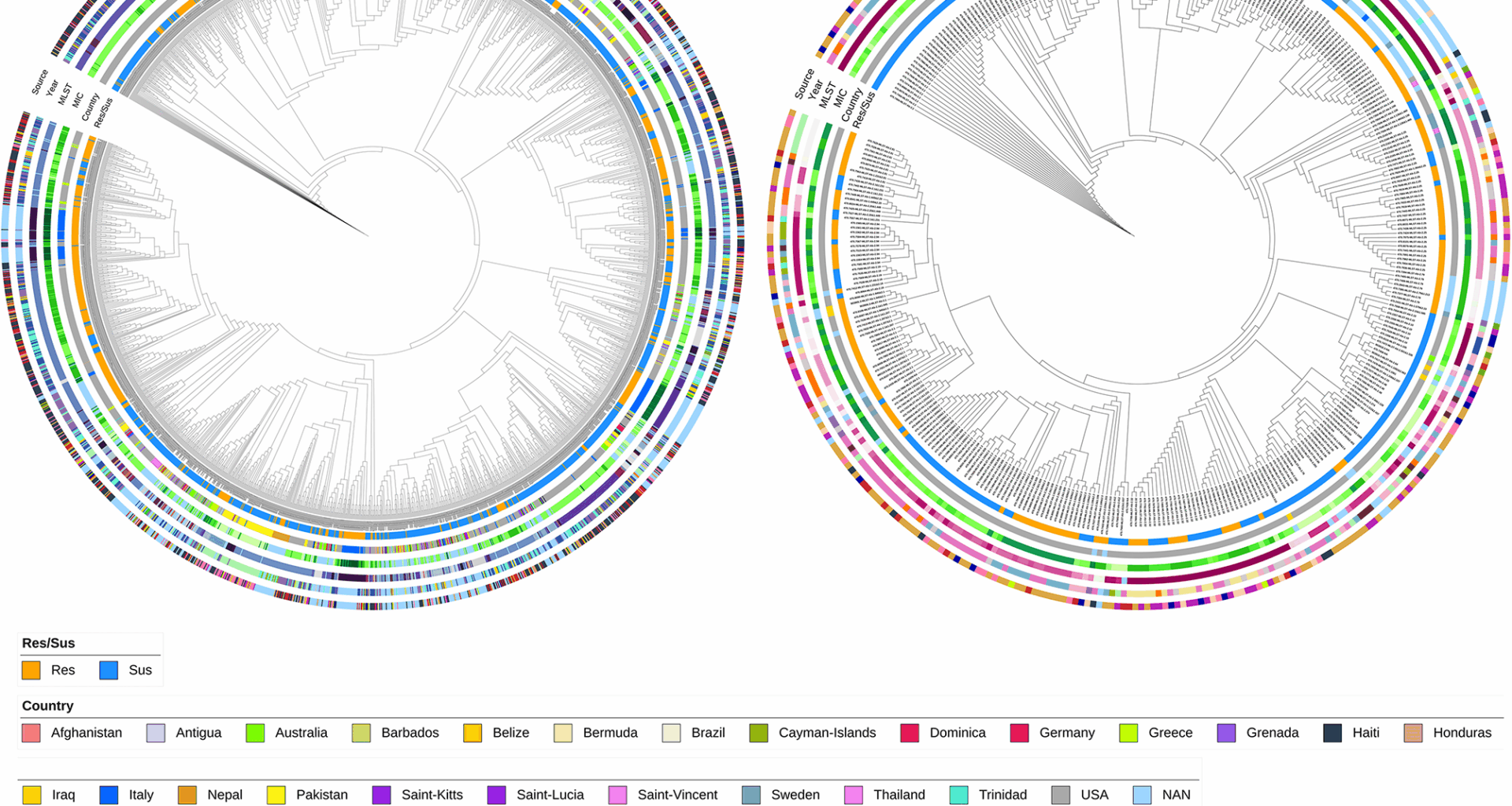
Genomic insights of KP and AB WGS data, which are resistant and susceptible to meropenem
A large-scale global genomic study was conducted to investigate the mechanism of meropenem resistance using 2,411 KP and 375 AB genomes from the BV-BRC database. Among KP, 959 isolates were resistant and 1452 susceptible; in AB, 192 were resistant and 183 susceptible (Table 1). AMR gene presence and missense mutations were profiled using the CARD database, identifying 321 AMR genes and 1579 AMR missense mutations in KP, and 184 AMR genes and 1119 AMR missense mutations in AB. Figure 1 depicts a feature-based phylogeny of all the genomes used in the study (Tables S1, S2), where resistant and susceptible isolates were interspersed across clades (Fig. 1 innermost circle). Country-specific clades were observed (23 countries for KP, 8 for AB), mostly from the USA (second innermost circle). The minimum inhibitory concentration (MIC) varied across clades: 0.12–128 mg/L in KP, 0.25–32 mg/L in AB (third circle). Most clades followed a consistent sequence type (ST): 215 STs in KP, 75 in AB (fourth circle from inside, Fig. 1a and b, and Figure S1). Samples ranged from 2008 to 2017 (KP) and 2003–2017 (AB) (second outermost circle). The collected isolates are from various sources and were broadly categorised into 10 different groups, with most from excretion (986, KP) and wounds (139, AB) sources (outermost circles, Fig. 1a and b).
Table 1 Overall dataset collected for the studyFig. 1
Phylogenetic tree of a. Klebsiella pneumoniae and b. Acinetobacter baumannii isolates were constructed using a presence-absence matrix of AMR genes and missense mutations. The concentric circles represent key metadata from the innermost to the outermost layer: (1) resistant/susceptible phenotype (2), country of isolation (3), minimum inhibitory concentration (MIC) values (4), multi-locus sequence typing (MLST) (shown in Fig. S1) (5), year of isolation, and (6) source of isolation
We categorized the AMR genes into six resistance mechanism categories defined by the CARD database: antibiotic inactivation, antibiotic efflux, target alteration, target protection, target replacement, and reduced permeability to antibiotics.
Table 2 provides the overall proportion of AMR genes categorised based on resistance mechanism in KP and AB. On highlighting some of the major entries, we found that when analysing AMR gene proportions, antibiotic efflux is the predominant resistance mechanism in both KP and AB. Separate analysis of meropenem-resistant and susceptible genomes revealed similar distributions of resistance mechanism classes with a slight increase in antibiotic inactivation observed in resistant isolates. AMR genes across different mechanism categories are listed in Tables S3 and S4.
Table 2 AMR genes and mutation percentages categorised based on resistance mechanisms
AMR gene mutation details indicate that approximately 32% of AMR genes undergo mutations in KP, whereas 27% in AB. Similarly, Table 2 highlights the proportion of missense mutations across resistance mechanisms, revealing notable differences in the mutation percentage of the antibiotic target alteration class and antibiotic efflux class between KP and AB. Overall mutation ratios between resistant and susceptible phenotypes were similar across resistance categories. Mutated AMR genes by mechanism category are listed in Tables S5 and S6.
To investigate the meropenem resistance mechanisms, we focused on key contributors: carbapenemases, efflux pumps, porins, and PBPs. Carbapenemases, part of the antibiotic inactivation class, play a central role by degrading carbapenems. Given their importance, we investigated the presence of carbapenemases in the collected isolates. Based on BLDB, we identified 16 carbapenemases in KP and 44 in AB (highlighted in Tables S3 and S4). In KP, carbapenemases included class A (e.g., blaGES and blaKPC), class D (blaOXA), and sub-class B1 (blaIMP, blaNDM, and blaVIM variants). blaKPC−2 and blaKPC−3 are especially prevalent. blaKPC−2 was found in 51% of the carbapenem-resistant genomes and 2% of the susceptible genomes, whereas blaKPC−3 was present in 29% of the resistant genomes and in only one genome from the susceptible group (highlighted in Table S3). In AB, sub-class B1 (blaNDM) and class D (blaOXA) carbapenemases were common, with a total of 43 blaOXA variants and blaNDM−1 identified. Among the carbapenemases, blaOXA−23 was detected in 59% of resistant and 1% of susceptible AB genomes, while blaOXA−66 appeared in 27% of resistant and 33% of susceptible genomes (as highlighted in Table S4). No carbapenemase mutations were observed in either species. We also examined other beta-lactamase genes such as carbenicillinase, broad-spectrum, narrow-spectrum, and ESBL genes. In KP, class A enzymes were most common (113 genes), followed by class C (7) and class D (6). In AB, class C beta-lactamases dominated (57 genes), followed by class A (11) and class D (3).
Importantly, non-carbapenemase carbapenem resistance has been previously reported in KP by Rosas N. et al., 2023. In our dataset, 67 (~ 7%) meropenem-resistant KP isolates lacked known carbapenemase genes, while 4% of the susceptible isolates carried at least one. These non-carbapenemase-resistant KP genomes contained 117 AMR genes and 215 missense mutations. Among other β-lactamase genes, we identified 27 class A, 3 class C, and 5 class D genes. Over 90% of these isolates carried resistance genes associated with antibiotic efflux (9 genes), target alteration (7 genes), reduced permeability (6 genes), and antibiotic inactivation (1 gene). Similarly, mutations were frequently observed in genes related to antibiotic target alteration (7 genes), efflux (4 genes), and reduced permeability (2 genes), each present in over 90% of the isolates. In contrast, all carbapenem-resistant AB isolates harboured at least one carbapenemase gene, whereas 98% of susceptible AB isolates also carried at least one.
Analysis of the antibiotic efflux category revealed 50 efflux-associated genes in KP and 27 in AB. In KP, efflux pumps from the RND (20), SMR (4), MFS (22), ABC (3), MATE (1) were observed. The most frequent efflux genes in KP include acrA (99.95%), kpnF (99.87%), lptD (99.71%), and kpnG (99.58%), along with efflux regulators such as acrR (99.21%), kpnE (97.92%), oqxA (97.22%), and oqxB (96.47%) (Table S3). Many of these genes harboured multiple mutations, with ramR (with 91 mutations), oqxB (90), oqxA (58), and lptD (57) among the most mutated, as reported in Table S5. Notably, mutations in acrA, kpnG, and crp appeared in over 90% of KP isolates (Table S5). In AB, efflux pumps from the RND (14), MATE (1), SMR (3), and MFS (9) families were identified. The RND efflux pumps, including adeI, adeJ, adeK, adeF, adeG, adeH, abuO, adeA, and adeB, along with their regulators, adeR, adeS, adeL, and adeN, the MATE family efflux pump abeM, the SMR family efflux pump abeS, and the MFS family efflux pumps amvA and abaF, are present in more than 90% of the isolates [37] (Table S4). The ade family genes (adeA, adeB, adeC, adeJ, adeH, adeS, adeF) and the amvA gene have more missense mutations. Common high-frequency mutations present in > 90% isolates included adeJ|Met1043Ile, abeM|Ser233Asn, adeN|ProPhe11HisIle, and the regulatory gene adeS|Lys339Gln (Table S6).
In terms of porins, KP carries 12 porin genes and AB has 3. In KP, highly prevalent porins include ompA and mdtQ (99.91%), ompK36 (99.41%), ompK35 (99.08%), ompK37 (98.79%), and lamB (96.93%) (Table S3). mdtQ harbours 103 missense mutations. Frequent mutations such as OmpA|Thr332Asn, OmpK37|Ile70Met, and OmpK37|Ile128Met occur in over 97% of KP isolates (Table S5). In AB, lpsB was the most common (98.66%), and multiple mutations were observed in the genes lpsB, oprD, and carO. Variants like LpsB|Ile157Val, LpsB|Gly146Asp, and oprD mutations (Asn411Asp, Thr415Val) are found in over 90% of AB isolates (Table S6).
Other AMR mechanism categories in the CARD include antibiotic target alteration, antibiotic target replacement, and antibiotic target protection. KP has 28 genes in target alteration, 15 in protection, and 22 in replacement categories. Seven alteration genes occur in > 99% of KP isolates, unlike protection or replacement genes (Table S3). Genes phoQ, arnT, rpoB, and eptB show over 50 mutations each. Penicillin-binding proteins (PBPs) are direct targets of carbapenems and fall under antibiotic target alterations. According to CARD data, we detected pbp3 in 99.91% of KP genomes, and it has 40 associated missense mutations. PBP-3|Val375Ala was the most frequent among them (Table S5). In AB, we identified five genes under alteration, one under protection, and six under replacement. Among which four of the alteration genes are present in > 98% of isolates, and none from other categories showed such prevalence. pbp3 is absent in AB, while parC and gyrA show multiple mutations (Tables S4 and S6).
ML/DL based identification of features contributing to meropenem resistance
Given the absence of any specific difference in the overall trend of AMR genes and mutation presence between the resistant and susceptible categories in both organisms, an ML/DL based classification approach was used to identify the significant features contributing to meropenem resistance. To identify significant features, we performed a chi-square test (p ≤ 0.05) on the full feature sets of 1900 for KP and 1303 for AB. This resulted in 410 and 211 significant features, respectively, encompassing AMR genes and missense mutations in KP and AB. With these features, we explored six machine learning models (LR, DT, RF, SVM, XGB, and MLP) and a DNN and the 5-fold cross-validation accuracy values are reported in Figure. S2. After optimization, the models were evaluated based on key performance metrics, including accuracy, precision, recall, and F1-score. The training and test metric values of all seven models in KP and AB are depicted in Fig. 2a and b, respectively. On the basis of these metrics, the SVM with a linear kernel model exhibited the best overall performance for both organisms, achieving test accuracy and F1-score values of 96% and 96% for KP and 93% and 93% for AB, respectively (Fig. 2a and b). Subsequently, sequential feature selection was performed using the SVM model to identify the most important features for differentiating resistant and susceptible phenotypes. This process led to the selection of seven features for KP and 10 for AB, which can discriminate between resistant and susceptible classes (Fig. 3a and c). When trained and tested using these selected features, the SVM model achieved accuracies of 94% and 92% on the training set (Fig. 3a and c) and 95% and 95% on the test datasets of KP and AB, respectively (Fig. 3b and d). To assess the model’s generalizability, external validation was performed using the selected features. The SVM model demonstrated robust performance with external validation accuracies of 95% for KP and 94% for AB (Fig. 3b and d).
Fig. 2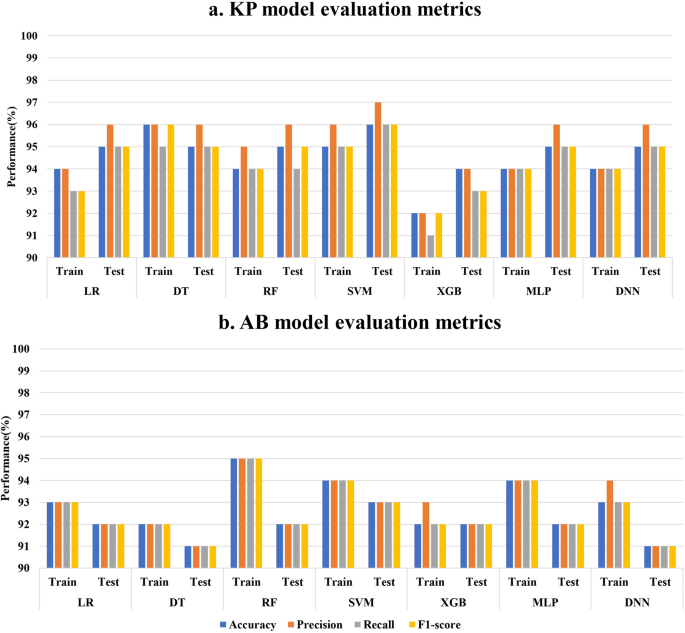
Performance metrics (accuracy, precision, recall, and F1-score) for six ML models and one DL model based on the significant feature set selected based on the chi-square test for training and testing datasets of a KP and b AB
Fig. 3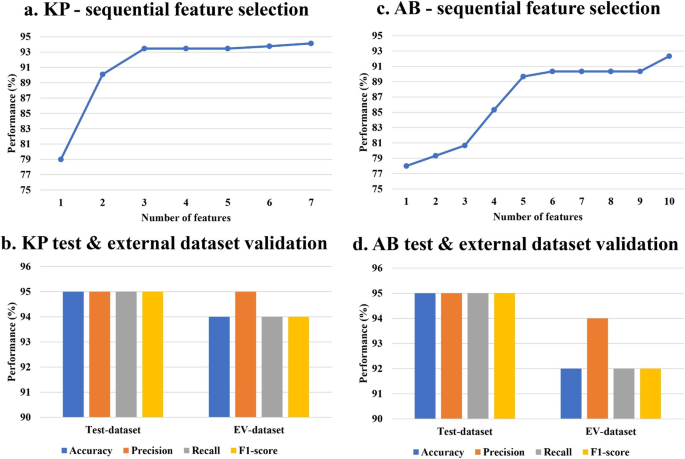
Sequential feature selection using the best-performing SVM model resulted in a 7 features for KP, reaching 94% training accuracy, and c 10 features for AB, reaching 92% training accuracy. The test dataset and the external dataset validation performed using the selected features yielded 95% & 94% accuracy in b KP and 95% & 92% accuracy in d AB
The seven selected key features of the KP are reported in Table 3. Among the AMR genes, carbapenemases blaKPC−2 and blaKPC−3, metallo-β-lactamase bleMBL, and aminoglycoside acetyltransferase aac(6’)-Ib9 were identified as key features. Three mutations were identified in genes such as oqxA, kpnE, and kpnG. The AMR genes, blaKPC−2, blaKPC−3, bleMBL, and aac(6’)-Ib were significant and were found predominantly in the resistant phenotype category, whereas the mutations oqxA|Lys308Gln, Kpne_KpnE|Ile96Val, and Kpne_KpnG|Ala69Ser were found significantly in genomes with the meropenem-susceptible phenotype (Table 3).
Table 3 Details of seven features identified from SVM-based feature selection for KP. The resistant and susceptible columns include both count and percentage
Table 4 lists the 10 key features identified for AB. The six AMR genes, blaOXA−23, blaOXA−113, blaOXA−174, blaOXA−312, blaOXA−72, and aac(6’)-Ib7, were identified as key features showing a significant count with the resistant phenotype (Table 4). Additionally, four mutations were identified in genes such as tet(A), adeR,oprD, and gyrA. Carbapenemases, including blaOXA−23, blaOXA−113, blaOXA−174, blaOXA−312, blaOXA−72 and the aminoglycoside acetyltransferase aac(6’)-Ib7, were predominantly found in the resistant category. The mutations, Abau_OprD_IPM|Asn411Asp, and Abau_gyrA_FLO|Ser81Leu, were mainly found in the resistant category, whereas tet(A)|Ala109Thr, and adeR|Leu142Ile, were found mainly in the susceptible category (Table 4). From the feature selection approach, we could identify the key features in KP and AB that can distinguish between the meropenem-resistant and susceptible classes. We further explored the association among the identified features contributing to the AMR phenotype.
Table 4 Details of 10 features identified from SVM-based feature selection for AB. The resistance and susceptible columns include both count and percentageAssociation rule mining of the selected features
To uncover potential hidden associations among the identified features of KP and AB, we performed association rule mining using seven selected features for KP and 10 selected features for AB. For KP, we identified 23 rules with a support value of 0.01 and a confidence value of 0.80 (Table S7). To identify the factors associated with meropenem resistance in KP , we filtered the rules to focus on those with a meropenem-resistant phenotype as the consequent. We obtained five association rules that are linked to the resistant phenotype with a lift greater than two, involving four features of the AMR genes blaKPC−2, blaKPC−3, and bleMBL, aac(6’)-Ib9. The network representation of the five association rules contributing to the meropenem-resistant phenotype depicted in Fig. 4a indicates that the four AMR genes are associated with the meropenem-resistant phenotype (Table S7). Association rules indicate that the presence of any one of blaKPC−2, blaKPC−3, bleMBL or aac(6’)-Ib9 can individually contribute to meropenem resistance. The aac(6’)-Ib9 gene is also found in combination with blaKPC−3 contributing to meropenem resistance (Table S7). We also identified seven rules involving mutations that contribute either individually or in combination to the meropenem-susceptible phenotype, with lift values ranging from 1.3 to 1.5. The three mutations were found to be in efflux pump genes.
Fig. 4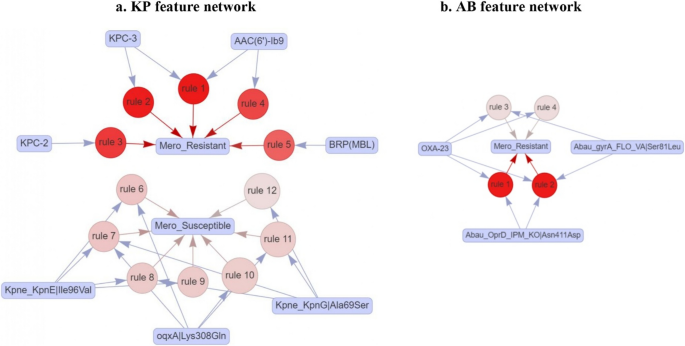
a Network of 12 association rules based on AMR genes and mutations in KP (antecedents) contributing to meropenem resistant and susceptible (consequents). b Network of 4 rules based on AMR genes and mutations in AB (antecedents) contributing to meropenem resistant. The red shading indicates lift values ranging from 1.3 to 2.5 in KP and 1.91 to 1.93 in AB
For AB, we identified 84 rules with a support value of 0.06 and a confidence value of 0.80 (Table S8). To pinpoint the factors contributing to meropenem resistance in AB, we filtered the rules to focus on those with a meropenem-resistant phenotype as the consequent, resulting in only four rules associated with the resistant phenotype. Among the 10 features used for association rule mining, three were found to be linked to the meropenem-resistant phenotype. These include one AMR gene (blaOXA−23) and two mutations (Abau_OprD_IPM|Asn411Asp, Abau_gyrA_FLO|Ser81Leu) with a lift of 1.91 to 1.93. The network representation of the association rules is shown in Fig. 4b. blaOXA−23 contributes individually to the meropenem-resistant phenotype. None of the mutations individually contributed to the resistant phenotype; they contributed to resistance in combination with blaOXA−23 (Table S8).
Analysis of antibiotic co-resistance and shared resistance patterns in KP and AB isolates
The isolates of KP and AB used in this study were found to be multi-drug resistant. Consequently, their genomes are expected to harbour features contributing to resistance against various antibiotics. To identify meropenem-associated co-resistance patterns with other antibiotics, we performed association rule mining using antibiotic resistance data available for the isolates. In KP, 24 different antibiotics from 9 classes were analysed. Using association rule mining with a support of 0.1, a lift value greater than 2, and a confidence value greater than 0.98, we identified 87 rules. Based on the analysis of antibiotic co-resistance, 18 antibiotics (ampicillin, ampicillin/sulbactam, aztreonam, cefazolin, cefepime, cefoxitin, ceftazidime, ceftriaxone, cefuroxime, ciprofloxacin, ertapenem, gentamicin, imipenem, levofloxacin, meropenem, piperacillin/tazobactam, tobramycin, and trimethoprim/sulfamethoxazole) from 7 different classes were found to be associated. The associated antibiotic classes in KP include carbapenems, cephalosporins, fluoroquinolones, monobactams, penicillins, aminoglycosides, and combination antibiotics (Fig. 5a).
Fig. 5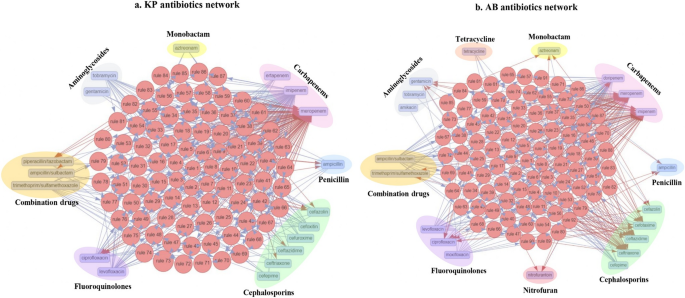
Network representation of shared and co-resistance among antibiotic classes. a Depicts 87 association rules in KP. b Depicts 92 association rules in AB
In AB, 24 antibiotics from 10 different classes were included in the study. Using association rule mining with a support of 0.1, a lift value greater than 2, and a confidence value greater than 0.98, we identified 92 rules. Based on the analysis of antibiotic co-resistance, 20 antibiotics (imipenem, meropenem, doripenem, ceftazidime, trimethoprim/sulfamethoxazole, ampicillin, ceftriaxone, moxifloxacin, cefepime, ampicillin/sulbactam, levofloxacin, nitrofurantoin, tetracycline, cefotaxime, ciprofloxacin, gentamicin, cefazolin, aztreonam, tobramycin, amikacin) from 9 different classes were found to be associated. The antibiotic classes associated with AB include carbapenems, cephalosporins, combination antibiotics, penicillin, fluoroquinolones, nitrofuran, tetracyclines, aminoglycosides, and monobactam (Fig. 5b).