
To discover schizophrenia risk genes, we generated exome-sequencing data on a new sample of 4650 cases and 5719 controls, and meta-analysed these data with RCVs from published sequencing studies of schizophrenia for a total of 28,898 cases, 103,041 controls and 3444 trios. This represents, to our knowledge, the largest whole-exome sequencing study of schizophrenia to date.
We report association at exome-wide significance between rare PTVs and damaging missense variants in STAG1 and schizophrenia. STAG1 encodes a subunit of cohesin, a protein complex required for correct chromosomal segregation during cell division18. Defects in cohesin subunits and interactors are associated with a heterogeneous class of neurodevelopmental disorders termed cohesinopathies, whose pathology is thought to be mediated by a further role of cohesin in 3D genome organisation19. Cohesin participates in both chromatin looping20 and formation of topologically associating domains (TADs)21. Studies have shown that loss of cohesin components, including STAG1, is associated with disrupted patterns of chromatin contact and gene expression, including genes with functions related to neuronal development22,23,24. Previous sequencing studies of schizophrenia have provided additional evidence for dysregulated chromatin in schizophrenia, by showing cases are enriched for RCVs and de novo coding variants in sets of genes related to chromatin modification and organisation12,25. Moreover, schizophrenia cases carry an excess of rare structural variants disrupting TAD boundaries compared with controls26. By implicating STAG1 at exome-wide significance in schizophrenia, we contribute further evidence suggesting an aetiological role for disrupted chromatin organisation in this disorder. In future studies, WGS can be used to investigate the role of TAD disrupting non-coding rare variants in schizophrenia, and also to determine whether schizophrenia is associated with disrupted TAD boundaries in specific cell types and/or developmental timepoints.
We also found that rare PTVs in ZNF136 are enriched in schizophrenia at exome-wide significance. ZNF136 encodes a zinc-finger protein that contains a Krüppel-associated Box (KRAB) domain, which is thought to act as a transcriptional repressor27,28. However, the functional roles of ZNF136 are not well characterised. Unlike all remaining genes currently shown to be enriched for PTVs in schizophrenia with exome-wide significance, ZNF136 displays no evidence for selective constraint against PTVs (gnomAD probability of being loss-of-function intolerant = 0)10. Small-scale transcriptomic studies suggest that ZNF136 is downregulated in schizophrenia29,30, but the mechanisms by which PTVs in ZNF136 may increase risk for schizophrenia are unclear.
It is important for exome-wide gene discovery studies to apply stringent genome-wide thresholds for statistical significance to reduce the reporting of false positives and to ensure that funding for functional follow-up studies is prioritised towards true targets31. STAG1 and ZNF136 were provisionally implicated by the SCHEMA study at FDR < 5%12, but our findings with a larger sample indicate that these genes reach exome-wide significance after Bonferroni correction for multiple testing. Other genes implicated in schizophrenia by the SCHEMA study, which did not achieve exome-wide significance after Bonferroni correction but which passed the FDR < 5% threshold, have also been implicated with greater certainty in larger samples13. In this regard, we also identified 6 additional genes associated with schizophrenia for the first time at FDR < 5% (SLC6A1, PCLO, ZMYND11, BSCL2, KLC1 and CGREF1). SLC6A1 and KLC1 are the first genes to be implicated in schizophrenia at FDR < 5% by missense variants (MPC > 2) alone. Sequencing data included in the published SCHEMA study supports association between damaging missense variants (MPC > 2) in these genes and schizophrenia12, but the SCHEMA analysis down-weighted association statistics for missense MPC 2-3 variants relative to PTVs and missense MPC > 3 variants, resulting in lower power to detect associations of this class12. SLC6A1 encodes a gamma-aminobutyric acid (GABA) transporter (GAT-1), which is highly expressed in GABAergic neurons and mediates uptake of GABA from the synaptic cleft of inhibitory synapses. Recently published in vitro GABA uptake assay data provides evidence that some of the published schizophrenia SLC6A1 missense variants, as well as two of the three missense variants reported in the new cases, confer loss-of-function effects on GAT-1 protein leading to reduced GABA uptake32. Thus, our study supports the hypothesis of haploinsufficiency being the disease mechanism underlying risk for schizophrenia from missense variants in SLC6A1. The other missense variant enriched gene, KLC1, encodes a light chain subunit of kinesin, a tetrameric protein complex responsible for intracellular transport along the cytoskeleton. Common schizophrenia risk alleles at the KLC1 locus are associated with reduced expression of KLC1 RNA transcripts in the human fetal brain33,34, and knockdown of KLC1 has been found to impair neuronal differentiation35; however, the functional impact of KLC1 missense variants observed in schizophrenia risk are unknown. A more detailed overview of the biological functions of the novel FDR < 5% genes reported in the current study is provided in Supplementary Note 2, and the spatio-temporal expression profiles of the novel exome-wide significant and FDR < 5% genes is provided in Supplementary Note 3.
Previous studies have identified four genes which show both fine-mapped common variant signals in schizophrenia GWAS and an excess of RCVs in cases at either exome-wide significance (GRIN2A and SP4) or FDR < 5% (STAG1 and FAM120A)4,12, providing strong evidence for their role in schizophrenia. Our study strengthens the evidence for STAG1. It also provides orthogonal support for the prioritisation of KLC1 as a credible causal gene underlying a complex GWAS signal at this locus. The convergence of common and rare genetic liability in STAG1 and KLC1 makes them attractive targets for researchers aiming to develop animal and cellular models of rare high-risk variants, as the common allele signal implies that mechanistic insights gained from these models may have broad relevance across cases. When examining RCV enrichment in genes impacted by schizophrenia risk CNVs, an excess of PTVs in NRXN1 was observed in cases compared with controls. This is a plausible finding, since intragenic deletions of NRXN1 have consistently been shown to increase risk for schizophrenia7,36. No genes overlapping multi-genic schizophrenia CNV loci were enriched for RCVs after correction for multiple testing.
Genes enriched for RCVs in schizophrenia often exhibit pleiotropic effects for other psychiatric and developmental disorders, particularly DD and ASD12,37. Four of the eight novel exome-wide significant and FDR < 5% genes identified in the current study show enrichment for RCVs in large sequencing studies of DD, ASD and epilepsy, providing orthogonal support for their role in schizophrenia. SLC6A1 has the broadest pleiotropic effects, wherein missense variants are enriched in ASD, DD and epilepsy. PTVs in SLC6A1 are also associated with DD. Several of the schizophrenia genes reported in the current study also demonstrate association with syndromic neurodevelopmental disorders; for example, PTVs, missense variants, and deletions in STAG1 cause a syndromic cohesinopathy characterised by developmental delay and mild dysmorphic features, sometimes accompanied by autistic traits and epilepsy38,39,40. Homozygous PTVs in PCLO are associated with pontocerebellar hypoplasia type III41, a cause of global developmental delay and seizures. Furthermore, loss and gain-of-function mutations in BSCL2 have been implicated in lipodystrophy and neuropathic conditions, respectively42. While many of the schizophrenia genes reported here show evidence of genic pleiotropy across psychiatric and developmental disorders, this does not imply functional or mechanistic overlap between these disorders, since different variants in the same gene can have distinct functional effects. Previous studies have provided evidence for pleiotropic effects from individual RCVs across schizophrenia, autism and developmental disorders37,43, however, demonstrating allelic pleiotropy for the schizophrenia genes reported here is beyond the scope of the current study. Future studies should also determine whether particular clinical features, including neurodevelopmental phenotypes, are enriched among schizophrenia cases carrying mutations in these pleiotropic genes.
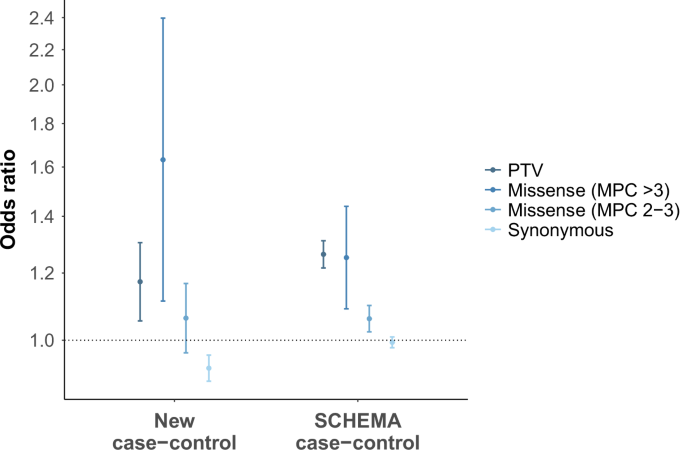
Gene-set analysis in the new sample of genes previously implicated in schizophrenia at exome-wide significance confirmed this set of genes is enriched for rare PTVs in cases compared with controls. While single-gene analysis in the new sample was underpowered, SETD1A, XPO4 and SRRM2 were enriched for RCVs in the new cases at nominal significance (P < 0.05). However, both our own study and a previous targeted sequencing study13 found higher rates of rare PTVs and damaging missense variants in CACNA1G in controls than in cases, suggesting further work is required to determine whether CACNA1G is a true schizophrenia risk gene. In the new sample, we found the rate of synonymous singleton variants in constrained genes to be lower in cases compared with controls. In the context of the low coding de novo mutation rate and a disorder for which damaging de novo coding variants are a risk factor, ascertainment of probands with that disorder will be enriched for those where the random occurrence of a de novo variant is a damaging nonsynonymous mutation rather than a synonymous one. The underrepresentation of singleton synonymous variants in the new cases may reflect this ascertainment bias rather than a true protective effect of synonymous variation. However, the large SCHEMA case-control analysis did not observe any difference in the rate of rare synonymous variants in constrained genes between cases and controls, and therefore a depletion of these variants in the new cases may also be a chance finding.
A strength of our study is the large number of newly exome-sequenced cases, which we meta-analyse with published data to increase power for gene discovery. Additionally, the inclusion of a missense only variant test identified two novel genes at FDR < 5% significance. Our study also has limitations. We lack deep and longitudinal phenotype data for most of the new and published cases included in our analysis, and we are therefore unable to determine whether variants in the novel genes reported are associated with particular clinical features. This limitation can be addressed by high-quality case reports for individuals carrying mutations in the genes implicated in our study, or by genomic studies with access to linked electronic healthcare data. Moreover, we are underpowered to analyse all genetically inferred population groups in the new sample. Increasing the diversity of sequenced samples in schizophrenia will both facilitate genomic discovery and ensure more equitable progress in precision psychiatry.
In conclusion, our study implicates STAG1 and ZNF136 in schizophrenia with exome-wide significance and 6 additional genes at FDR < 5%. Many of these genes are enriched for RCVs in DD, ASD, and epilepsy, which supports their association with schizophrenia given the known genetic overlap between these disorders. We strengthen the evidence for an allelic series of common and rare schizophrenia risk alleles in STAG1, and provide evidence for the convergence of common and rare risk alleles in KLC1. Association of STAG1 at exome-wide significance provides further support for an aetiological role of disrupted chromatin organisation in schizophrenia, while association of SLC6A1 at FDR < 5% furthers the evidence implicating perturbed GABAergic neuronal signalling in the disorder.