
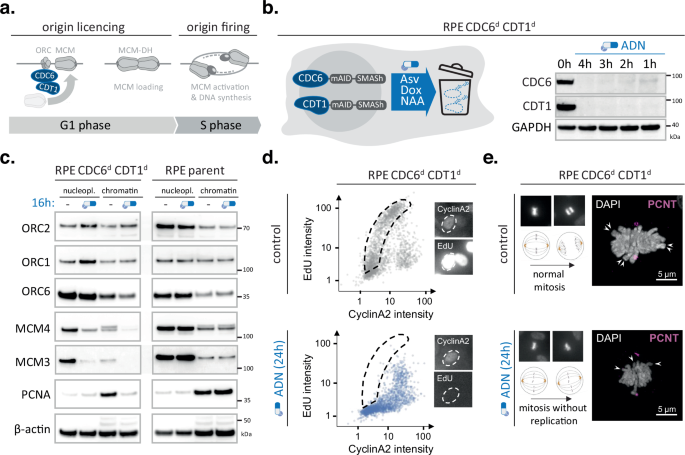
Turning off origin licensing within one cell cycle
We have previously developed protein depletion technologies to suppress initiation of DNA replication in human cells22. To fully inhibit DNA replication, we previously had to simultaneously block origin licensing (by CDC6 degradation) and origin firing (by CDC7 inhibition)22. Since licensing and firing are fundamentally distinct stages of replication initiation and CDC7 has possible confounding roles in checkpoint signalling23, we generated a human cell model exclusively targeting origin licensing. To achieve high efficacy and temporal resolution within a single G1 phase, our model combines two synergistic degron tags, SMASh and mAID, and expression of a recently identified co-factor ARF-PB1 that improves degradation dynamics22,24. The SMASh tag is a large self-cleaving peptide that rapidly removes itself from the protein of interest; upon addition of the small molecule Asunaprevir (ASV) self-cleavage is blocked, rendering newly translated fusion proteins unstable and/or dysfunctional (Supplementary Fig. 1a). At the same time, the mAID tag allows active recruitment of an ectopic E3-ligase (OsTIR) and upon addition of a small synthetic auxin molecule (NAA) triggers rapid ubiquitination and proteasomal degradation of the remaining target proteins (Supplementary Fig. 1a). To prevent constitutive degradation of mAID-tagged proteins and further improve protein degradation kinetics, we introduced a co-expression construct that allows doxycycline-(DOX) inducible expression of OsTIR and the PB1 domain of ARF16 (ARF-PB1) from the human Rosa26 safe harbour locus (Supplementary Fig. 1A). Using CRISPR-based genome-editing, we introduced this system into human RPE1 p53-/- cells and tagged both alleles of endogenous CDC6 and CDT1 with a mAID-SMASh double-degron. The resultant RPE Cdc6d Cdt1d cells showed rapid depletion of both target proteins – with levels of both full-length proteins being very low and undetectable at 1 and 4 h post-induction, respectively (Fig. 1b). CDC6 and CDT1 are both required for ORC to load the MCM helicase on DNA. Selective degradation of these proteins should, therefore, reduce MCM loading on chromatin without affecting ORC (Fig. 1a). Indeed, exposing RPE Cdc6d Cdt1d cell to the degron drugs ASV/DOX/NAA (ADN) for 16 h eliminated MCM4 and MCM3 from the chromatin fraction, while preserving ORC1 and ORC2 (Fig. 1c). Parallel treatments using the parental RPE1 p53-/- cells did not alter chromatin-bound MCM or ORC levels, verifying that the ADN treatment, by itself, did not compromise licensing (Fig. 1c).
To study how simultaneous depletion of CDC6 and CDT1 alters DNA replication and cell cycle progression, we performed quantitative image-based cytometry (QIBC) of large cell populations and for each cell plotted 5-Ethynyl-2′-deoxyuridine (EdU) nucleotide incorporation levels versus nuclear Cyclin A2 intensities. Asynchronous, mock-treated RPE Cdc6d Cdt1d cells showed a typical arc-shaped distribution, indicative of efficient DNA replication initiation upon S-phase entry when Cyclin A2 levels start to accumulate (Fig. 1d). In contrast, ADN-treated RPE Cdc6d Cdt1d cells failed to trigger high EdU incorporation rates in early S phase, while allowing CyclinA2 accumulation, revealing a strong and selective defect in DNA replication initiation in the absence of CDC6 and CDT1 (Fig. 1d). Independent FACS experiments confirmed a complete block of DNA replication upon ADN treatment, which relied on degron-tagged CDC6 and CDT1 (Supplementary Fig. 1b). Notably, depleting CDC6 and CDT1 prevents the production of DNA replication forks, and thus is fundamentally different from stopping DNA replication by impeding fork progression, which is known to cause DNA replication stress, DNA damage checkpoint activation and cell cycle arrest25. Directly comparing degradation of CDC6 and CDT1 with classical fork stalling agents such as hydroxyurea (HU) indicates that i) the RPE Cdc6d Cdt1d cells are proficient in DNA damage signalling and ii) loss of origin licensing does not trigger DNA replication stress markers such as H2AX or RPA hyperphosphorylation (Supplementary Fig. 2a). In fact, depletion of CDC6 and CDT1 suppressed spontaneous and HU-induced DNA damage, as expected in the absence of replication intermediates (Supplementary Fig. 2a). Moreover, since it did not prevent Cyclin A2 accumulation and mitotic entry, it led to mitotic cells without apparent sister-chromatids (Fig. 1e, Supplementary Fig. 1c and Supplementary Fig. 2b).
The ability to rapidly deplete CDC6 and CDT1 allowed us to ask whether origin licensing can occur any time during the G1 phase of the cell cycle. RPE Cdc6d Cdt1d cells, treated with nocodazole, were collected by mitotic shake-off, plated in fresh media and allowed to proceed through G1 into S phase. With this protocol, the cells entered S phase between 12 and 14 h after mitotic exit (Fig. 2a). To determine when origins are licensed during G1, the ADN drug mixture was added to the cells either during mitosis and removed at various time points in G1 or was added at various time points in G1 and kept until the 14 h time point after mitotic exit, at which time the fraction of EdU-positive cells was determined by flow cytometry. ADN reduced the number of EdU-positive cells in a treatment duration-dependent manner and, independent of whether the cells were exposed to ADN in the first or second half of G1 (Fig. 2b). These results suggest that origin licensing can occur in a cumulative manner throughout the G1 phase, consistent with previous reports26,27.
Fig. 2: CDC6 and CDT1 are needed throughout G1 phase to establish origin activity genome-wide.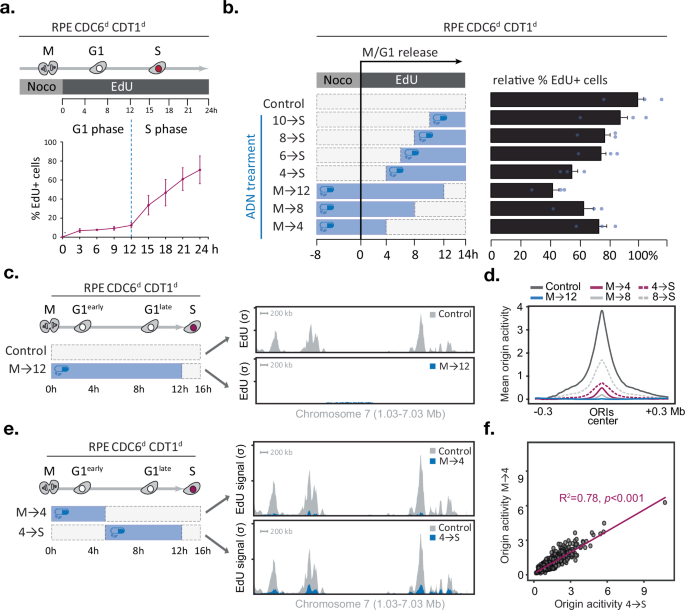
a–f CDC6 and CDT1 are required in early and late G1 phase to promote DNA replication initiation genome-wide, a Experimental outline (top) and line graph (bottom) depicting the mean percentage of EdU-positive cells after mitotic release determined by FACS (n = 3 technical replicates, error bars indicate SD). b Experimental outline (left) and bar graph (right) depicting the mean percentage of EdU-positive cells 14 h after mitotic release for each condition (n = 3 technical replicates, error bars indicate SD). CDC6 and CDT1 are needed in early and late G1 phase to promote origin activity; c Outline of the experiment and corresponding replication initiation profiles (EdUseq-HU) at a representative genomic region in mock (grey) or ADN-treated (blue) cell populations and collected 16 h after mitotic shake-off. Bin resolution, 10 kb; scale bar, 200 kb; σ, sigma (normalised number of sequence reads per bin divided by its SD), lower tick σ = 100, higher tick σ = 200. d Average origin activity (i.e. mean σ values) at 1 Kb resolution around 1000 predefined, most active early S-phase origins after different ADN treatments (as outlined in b), e Outline of the experiment and corresponding replication initiation profiles (EdUseq-HU) at a representative genomic region in mock (grey) or ADN-treated (blue) cell populations and collected 16 h after mitotic shake-off. Bin resolution, 10 kb; scale bar, 200 kb; σ, sigma (normalised number of sequence reads per bin divided by its SD), lower tick σ = 100, higher tick σ = 200. f scatter plot comparing EdUseq-HU (σ) values at 1000 individual early S-phase origins after treatment M→4 or 4→S (as outlined in e). Linear regression fit (purple line) with coefficient of determination (R²) indicating the proportion of variance explained between the two datasets; slope significance determined using a two-tailed t-test. No adjustment for multiple comparisons was performed. Source data are provided as a Source Data file.
While restricting origin licensing to specific G1 time windows did not prevent entry into S phase, the effect on the firing of individual origins could not be determined by the flow cytometry analysis. Moreover, the flow cytometry analysis classified cells as EdU-positive or EdU-negative but did not provide a quantitative assessment of the level of DNA replication initiation. We, therefore, resorted to a more detailed analysis of initiation of DNA replication by employing EdUseq, a method that can map origin firing in a genome-wide manner26,28. In line with the flow cytometry data, active degradation of CDC6 and CDT1 for 12 h throughout G1 (treatment M→12) abolished EdU incorporation at origins, as seen by inspecting a representative genomic region (chromosome 7, Fig. 2c) or by plotting the average EdUseq signal of the thousand most efficient origins (Fig. 2d).
Shorter treatments with ADN resulted in partial suppression of origin firing with the magnitude of the effect correlating with the length of ADN treatment. Thus, ADN treatments during the first 4 or 8 h of G1 phase (treatments M→4 and M→8) led to 85% and 94% reductions in average origin activity, respectively (Fig. 2d, e). Similarly, ADN treatments during the last 4 or 8 h of G1 phase (treatments 8→S and 4→S) led to 60% and 78% reductions in average origin activity, respectively (Fig. 2d, e). These results further indicate that ADN treatments suppressed origin firing when given either at the beginning or end of G1 phase.
To study if certain origins are licensed in early versus late G1 phase, we compared for each origin its EdUseq signal in the cells treated with ADN for the first 4 h of G1 (treatment M→4) versus the cells treated with ADN for the last 8 h of G1 (treatment 4→S). The comparisons demonstrated a highly significant correlation of signal intensities (R2 = 0.78) indicating that the majority of origins are impacted similarly by the two ADN treatments (Fig. 2e, f). A similar conclusion can be reached by examining the EdUseq origin firing data for all the other ADN treatments described above (Supplementary Fig. 3). Together, these data indicate that efficient DNA replication initiation requires MCM loading activities throughout G1 phase and that there is no preference for specific origins to be licensed early versus late in the G1 phase of the cell cycle.
CDK4/6 inhibition stalls origin licensing through RB
Given the key role of CDK4/6 activity in G1/S phase transition, and the fact that the expression profile of its activating partner Cyclin D1 mirrors the timing of MCM loading27,29,30,31, we wondered if CDK4/6 activity could be the missing link between replication licensing and cell cycle control. Selective CDK4/6 inhibitors cause a potent, yet fully reversible, G1 phase arrest, and thus are extensively used to obtain synchronous cell cultures32,33. Alternatively, the plant amino acid mimosine can be used to cause a reversible G1/S phase arrest34,35. Mimosine effectively stalls human cells before the onset of DNA replication, hence creating a synchronised population with maximum levels of loaded MCM (Supplementary Fig. 4a). To test if CDK4/6 activity is required for efficient origin licensing, we subjected RPE1p53-/- cells to mimosine and/or two independent CDK4/6 inhibitors, palbociclib or Trilaciclib17,34 and compared chromatin-associated MCM2 and CDC45 levels in fractionated lysates (Fig. 3a). Treatment with palbociclib or Trilaciclib diminished the levels of chromatin-associated MCM2 compared to mimosine or mock-treated controls. Adding palbociclib prior to mimosine reduced the levels of chromatin-associated MCM2 compared to mimosine-only controls, suggesting that CDK4/6 inhibition arrested cells in a distinct G1 phase state with incomplete origin licensing (Fig. 3a). Indeed, palbociclib or Trilaciclib arrested cells with increased Cyclin D1 and reduced Cyclin A2 levels, indicative of a G1 phase arrest prior to APC/C inactivation, while mimosine treatment caused elevated Cyclin A2 and reduced Cyclin D1 levels, indicative of an arrest at the G1/S phase transition post APC/C inactivation (Fig. 3a and11,34). Notably, the changes in chromatin-associated MCM2 are paralleled with changes in RB phosphorylation in the nucleoplasm fractions, substantiating a link between CDK4/6 activity and MCM loading efficacy (Fig. 3a). Follow-up studies using p53-proficient and p53-deficient epithelial cells, as well as primary human fibroblasts, confirmed that palbociclib limits MCM loading and indicated that this defect does not rely on p53 status (Supplementary Fig. 4b). Moreover, independent high-content imaging data confirmed a 4-5-fold decrease in chromatin-associated MCM2 and MCM6 in G1 cells upon palbociclib addition (Fig. 3b).
Fig. 3: CDK4/6 inhibitors prevent the completion of origin licensing via the RB protein family.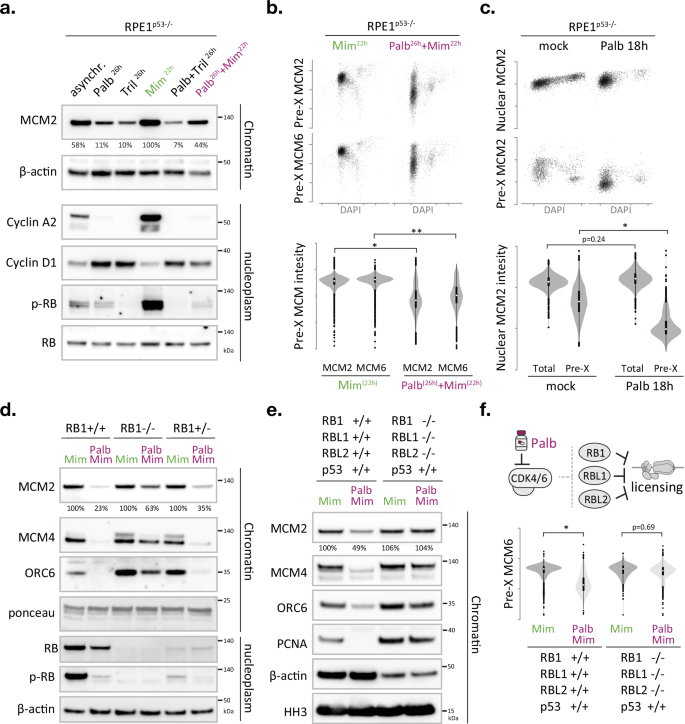
a–f The CDK4/6-RB axis controls origin licensing proficiency. a Immunoblot monitoring the effect of CDK4/6 inhibitors and/or mimosine on chromatin-bound MCM2 levels and corresponding cell cycle markers in nucleoplasm fractions. RB total and β-actin served as loading control (see Supplementary Fig. 4b) Representative blots are shown, with the experiment repeated twice. Values under immunoblots represent relative percentage of MCM2 signal compared to the Mimosine positive control. b QIBC analysis monitoring chromatin-bound MCM2 or MCM6 levels in single cells, relative to DNA content (DAPI), after expose to mimosine only or palbococlib and mimosine (analogous to the treatments in a). Scatterplots show MCM2/MCM6 levels in >1000 pre-extracted nuclei per condition. Violin plots depict distribution of MCM2/MCM6 signals in pre-extracted G1 phase nuclei (classified based on DAPI). The box shows the interquartile range (IQR) from 25th (Q1) to 75th (Q3) percentile; the white dot in the box corresponds to the median. Whiskers extend to the minimum and maximum values within 1.5 × IQR from Q1 and Q3, and dots outside this range represent potential outliers. Asterisks indicate a significant disparity between population means; *p = 0.0007; ** p = 0.0003; two-tailed paired t test; n = 4 technical replicates. c QIBC analysis monitoring total nuclear or chromatin-bound MCM2 levels in single cells, relative to DNA content (DAPI), after palbococlib expose. Scatterplots show MCM2 levels in >1000 nuclei per condition. Violin plots depict distribution of MCM2 signals in G1 phase nuclei (classified based on DAPI). The box shows the interquartile range (IQR) from 25th (Q1) to 75th (Q3) percentile; the white dot in the box corresponds to the median. Whiskers extend to the minimum and maximum values within 1.5 × IQR from Q1 and Q3, and dots outside this range represent potential outliers. Asterisk indicates a significant disparity between population means, p = 0.023; two-tailed paired t test; n = 4 technical replicates). d Immunoblot examining the effect of RB1 deficiency on palbociclib-induced licensing defects. The changes in chromatin-bound MCM2 are mirrored by chromatin-bound MCM4 and ORC6. Total RB protein and RB (S807/811) phosphorylation status is verified in nucleoplasm fractions. Ponceau S staining of bulk histones (10–25 kDa) and immunodetection of β-actin served as loading control. Nucleoplasmic CDC6 and CDT1 levels are shown in Supplementary Fig. 6a. e Immunoblot monitoring the effect of palbociclib and/or mimosine on origin licensing in RPE cells lacking all RB pocket proteins. d, e Cells were treated as in Fig. 4b and Supplementary Fig. 4b. Nucleoplasmic CDC6, CDT1 and RB phospho-RB (S807/811) phosphorylation levels are shown in Supplementary Fig. 6b. f QIBC analysis monitoring chromatin-bound MCM6 levels in single cells treated as in (e). Scatterplots show MCM6 levels in >1000 pre-extracted nuclei per condition. Violin plots depict distribution of MCM6 signals in pre-extracted G1 phase nuclei (classified based on DAPI). The box shows the interquartile range (IQR) from 25th (Q1) to 75th (Q3) percentile; the white dot in the box corresponds to the median. Whiskers extend to the minimum and maximum values within 1.5 × IQR from Q1 and Q3, and dots outside this range represent potential outliers. Asterisk indicates a disparity between population means; n = 5 technical replicates per condition, p = 0.00001 (two-tailed paired t test). Asynchr. asynchronous, Palb palbociclib, Tril Trilaciclub, Mim Mimosine, pre-X preextracted/chromatin bound. Mimosine only and Minosine+Palbociclib treatments are highlighted with green and purple labels, respectively, and performed as depicted in Supplementary Fig. 4b. Source data are provided as a Source Data file.
Due to its selective and reversible nature, CDK4/6 inhibition has become a widespread and recommended approach to synchronise cells32,33, however, long-term CDK4/6 inhibition (>3 days) should be prevented as it causes MCM complex instability, asynchronous releases and DNA damage36. We indeed find that long-term palbociclib and mimosine treatments (4–7 days) cause a stark drop in MCM2 levels in total cell lysates, while short treatments (1–2 days) do not significantly alter total MCM2 levels (Supplementary Fig. 4c). Adding the proteasome inhibitor MG132 to palbociclib and mimosine treated cells increases CDC6 and CDT1 expression but does not alleviate the MCM loading defect, suggesting that CDC6 or CDT1 protein levels are not limiting in this context and targeted MCM degradation is not the leading cause of the observed licensing defect (Supplementary Fig. 4d).
To directly compare the immediate effects of CDK4/6 inhibition on total and chromatin-bound MCM proteins (independent of mimosine), we exposed cells to palbociclib (200 nM) for 18 h and performed high-content imaging on pre-extracted and non-pre-extracted cells and quantified integrated MCM2 intensities in individual G1 phase cells. While 18 h palbociclib treatment did not significantly affect the mean intensity of total nuclear MCM2, it reduced chromatin-bound MCM2 more than 4-fold (Fig. 3c). Independent experiments in U2OS and RPE1 cells confirmed that a short pulse of Palbociclib (200 nM, 8-h) is sufficient to reduce chromatin-bound MCM2 levels in G1 phase nuclei (Supplementary Fig. 4e). We thus conclude that CDK4/6 inhibition causes a potent and immediate stall in origin licensing, which occurs before MCM protein levels become limiting.
CDK4/6 regulates cell cycle commitment by phosphorylating the RB family of pocket proteins, which modulates their binding to the E2F transcription factors that drive G1/S phase transition17. Interestingly, RB proteins bind multiple DNA replication initiation factors, such as ORC137, MCM738 and BRD421. Previous in vitro studies have indicated that human CDK activities evict RB from the ORC1-CDC6 complex while preserving the interaction between ORC1 and CDC639. To test if CDK4/6 inhibitors block licensing via RB, we obtained isogenic RPE1 clones40 with either normal levels of RB, no detectable RB or severely reduced levels of RB, and exposed these clones to palbociclib and mimosine, or mimosine alone, and determined the relative amount of chromatin-bound MCM2 and MCM4. RB deficiency alleviated the MCM loading defect imposed by palbociclib, directly implicating the tumour suppressor RB in origin licensing regulation (Fig. 3d and Supplementary Fig. 5a). The fact that the rescue is not complete and a relatively low dose of palbociclib (200 nM) can still reduce the level of chromatin-bound MCM in the absence of RB indicates that additional CDK4/6 targets exist that limit MCM loading in human cells. Humans have two additional RB-like proteins, RBL1/p107 and RBL2/p130, and overexpression of the latter is reported to impede DNA synthesis in a Xenopus in vitro replication assay38. We therefore investigated the effect of palbociclib in human RPE1 cells deficient for all three RB genes and found that the palbociclib-induced MCM loading defect is completely restored in RB1/RBL1/RBL2 triple knockout cells41 (Fig. 3e and Supplementary Fig. 5b). These experiments also revealed that palbociclib impaired ORC6 recruitment in an RB-dependent manner, supporting a key upstream role for CDK4/6 in origin licensing (Fig. 3d-e).
Given that RB loss is a well-established mechanism of resistance to CDK4/6 inhibitors in breast cancer patients, we wished to verify these findings using independent breast cancer models. A direct comparison of an RB-deficient (HCC1937) and two RB-proficient (MCF7 and MDA-MB-231) epithelial breast cancer cell lines substantiated the close correlation between RB (S807/811) phosphorylation and origin licensing efficacy (Supplementary Fig. 5c). These data also confirmed that Palbociclib effectively stalls the chromatin-recruitment of MCM2, MCM4 and ORC6 in RB-proficient cells and that this effect is alleviated in RB-deficient cells (Supplementary Fig. 5c).
Recent efforts to tackle therapy resistance involve proteolysis targeting chimeras (PROTACs)—small molecule drugs that trigger selective degradation of proteins in vivo. Notably, PROTACs degrading CDK4/6 have shown greater efficacy than traditional CDK4/6 kinase inhibitors in RB1-deficient cancer models, yet the mechanism behind this increased effectiveness remains to be clarified42,43. We find selective PROTACs targeting CDK4 (BSJ-04-132) or CDK6 (BSJ-03-123) to arrest human RPE1 cells in an under-licensed state (Supplementary Fig. 6a-d). While combined CDK4 and CDK6 PROTAC treatment and Palbociclib treatment were equally effective in stalling MCM loading (Supplementary Fig. 6c-d), we observed several notable differences at the molecular level. First, while the PROTACs reduced CDK4/6 protein levels, the catalytic inhibitor Palbociclib increased CDK4/6 protein levels (Supplementary Fig. 6a). Second, Palbociclib inhibited RB phosphorylation more effectively than the combined treatment with CDK4 and CDK6 PROTACs, even when the PROTACs were used at higher concentrations to compensate for potential reductions in catalytic site activity (Supplementary Fig. 6a)43. These findings support a model in which (i) both CDK4 and CDK6 promote cell cycle progression and MCM loading in human cells, and (ii) the redundant CDK activities phosphorylate RB in absence of CDK4/6 proteins. These results also confirm the strong concordance between S/G2 phase entry (Supplementary Fig. 6b) and origin licensing proficiency (Supplementary Fig. 6c-d), indicating molecular coupling between cell cycle and DNA replication commitment. We propose that CDK4/6 activity counteracts pocket proteins in G1 phase to coordinate origin licensing and cell cycle commitment.
RB blocks origin licensing independent of E2F-driven CDC6, CDT1 or MCM6 expression
A key role of CDK4/6-RB axis is to regulate E2F-dependent transcription17. Because CDC6, CDT1, and MCM6 are considered direct E2F-targets44, we investigated whether the expression of these proteins becomes limiting following treatment with Palbociclib. While we found nuclear CDC6 and CDT1 levels to be higher in RB knockout cells compared to their RB-proficient controls (as predicted for E2F target genes), we noted that the Palbociclib-induced changes in nuclear CDT1 and CDC6 expression did not correlate with the changes in origin licensing (Fig. 3d-e and Supplementary Fig. 5a-c). We found Palbociclib treatment to increase nuclear CDT1 levels while reducing licensing, suggesting that transcriptional control of CDT1 is not limiting in this context. While Palbociclib treatment reduced nuclear CDC6 levels, it did so in an RB-independent manner (Fig. 3d-e and Supplementary Fig. 5a-c), suggesting that the effect of CDK4/6 activity on CDC6 expression/protein stability and its effect on MCM loading (which is RB-dependent) can be uncoupled.
Earlier work by the Meyer and Diffley laboratories indicated that human CDK2 activity is needed in G1 phase to promote the transcription and protein stability of CDC69,12. To test if CDC6 expression is limiting for origin licensing upon CDK4/6 inhibition, we made use of a human bronchial epithelial cell model that allows inducible, E2F-independent expression of CDC6 (HBEC CDC6 Tet-ON)45. While doxycycline treatment caused significant CDC6 overexpression in all cell cycle stages (Supplementary Fig. 7a), it did not rescue the palbociclib-dependent origin licensing defect in HBEC CDC6 Tet-ON cells (Supplementary Fig. 7b). QIBC and biochemical analysis confirmed that palbociclib impaired MCM2, MCM4 and ORC6 loading (Supplementary Fig. 7b, c). Notably, CDC6 chromatin occupancy was not impaired by CDK4/6 inhibition, revealing that human CDKs regulate origin licensing post CDC6 recruitment and beyond CDC6 expression and/or protein stability (Supplementary Fig. 7d). To determine whether CDT1 levels could be limiting in this context, we transfected HBEC CDC6 Tet-ON cell with a human CDT1 overexpression construct and found that elevated CDT1 levels did not restore MCM2 loading in cells treated with palbociclib and doxycycline (Supplementary Fig. 7e). These observations are in line with origin licensing not being restored when CDT1 and CDC6 are stabilised by proteasome inhibition (Supplementary Fig. 4d) and the fact that nuclear CDT1 levels do not correlate with impaired origin licensing upon CDK4/6 inhibition (Supplementary Fig. 5a-c). We conclude that short-term CDK4/6 inhibition can block origin licensing in cells where expression of the MCM loaders CDC6 and CDT1 is not limiting.
Since MCM6 is a validated E2F target with multiple E2F binding elements in its promoter46, we directly compared total and chromatin-bound MCM6 levels upon Palbociclib exposure in RPE1 cells. QIBC analysis revealed that 18 h palbociclib treatment caused a stark reduction in chromatin-bound MCM6 in G1 phase cells without changing the total levels of MCM6 in G1 nuclei (Supplementary Fig. 5d). Parallel quantifications of MCM2 in these cells revealed identical results (Supplementary Fig. 5d). To further study the impact of the RB-E2F axis in licensing control, we transiently complemented RB1-deficient RPE1 cells with wildtype RB (RBwt) or mutant RB defective in E2F binding (RB661W)47 and found that both RB constructs significantly stalled MCM loading in Palbociclib-treated G1 cells (Supplementary Fig. 8a, b), further supporting a model where RB pocket proteins block origin licensing independent of E2F-driven transcriptional regulation. In Xenopus extracts, Rb and p130 impede DNA synthesis via a direct interaction with the C-terminal domain of MCM7 (MCM7-CT)38. We find that ectopic expression of MCM7-CT in human RPE1 cells prevents both RB phosphorylation and chromatin-recruitment of MCM2, substantiating a functional link between RB regulation and origin licensing (Supplementary Fig. 9).
CDK4/6 inhibitor-induced licensing defects can be perpetuated to cause mitosis with unreplicated DNA
Human cells license many more origins in G1 phase than are fired in an unperturbed S phase48, so we wondered if the observed reduction in ORC6 and MCM loading upon CDK4/6 inhibition also affected the pool of origins required for genome replication. The non-linear correlation between MCM detection and origin activity as well as the fact that CDK4/6 controls S-phase entry, make this a challenging question to address, but our biochemical analysis showed encouraging signs that the relevant pool of MCM complexes was affected. While RB deficiency partially restored MCM4 loading in cells treated with mimosine and palbociclib, we noted an apparent lack of the upshifted MCM4 band compared to the mimosine-only control, implying that CDK4/6 inhibition alters the nature of the loaded MCM complexes and/or their interaction with DDK (Fig. 3d). Recent cryo-EM studies have demonstrated that DDK requires MCM complexes to be in a double-hexameric state to be able to phosphorylate MCM2 and MCM4, which is the key step towards origin firing49,50,51,52. If CDK4/6 activity would be needed to load the pool of MCM complexes that drive DNA synthesis and act upstream of the MCM loading factors CDC6 and CDT1, one would expect cells to be sensitive to CDC6 and CDT1 depletion upon CDK4/6 inhibitor release. To test this hypothesis, we made use of our RPE Cdc6d Cdt1d cells and devised a quantitative IF setup that allowed us to rapidly block MCM loading upon palbociclib release and study cell cycle progression and DNA replication at single-cell resolution (Fig. 4a). If licensing did not occur during the transient palbociclib arrest, subsequent loss of CDC6 and CDT1 should sustain the licensing defect but allow S-phase entry. In line with this hypothesis, we found that palbociclib release in the presence of CDC6 and CDT1 led to efficient DNA replication, while palbociclib release upon CDC6 and CDT1 degradation led to a major drop in DNA synthesis (Fig. 4b, c). Combining palbociclib release with CDC6 and CDT1 degradation diminished EdU incorporation to undetectable levels in Cyclin A2-positive mid-S-phase cells, demonstrating that the majority of origins in CDK4/6 inhibited cells still require MCM loading activities in order to drive genome-wide DNA synthesis. Notably, the lack of EdU incorporation was not due to a failed drug release/persistent G1 arrest, since the CDC6 and CDT1-depleted cells did accumulate Cyclin A2 and ultimately entered mitosis (Fig. 4b, d). The proficiency in mitotic entry allowed us to independently confirm the failure to initiate DNA replication, as cytological analysis revealed abundant small DAPI-stained metaphase plates and a significant reduction in anaphase figures in cells depleted of CDC6 and CDT1 upon palbociclib release (Fig. 4d). Analogous assays using RL5a, a small molecule hampering ORC-DNA interactions53, confirmed these results and indicated that palbociclib impairs replication independently of our Cdc6d Cdt1d model and that combined CDK4/6 and origin licensing inhibition can be used to effectively trigger aberrant mitosis in p53-deficient cells (Supplementary Fig. 10a, b). The same palbociclib/RL5a combination treatments triggered a potent interphase arrest in p53-proficient cells, confirming that human RPE1 cells have a functional origin licensing checkpoint54,55. Biochemical analysis of the p53-deficient cells validated the uncoupling of DNA replication and cell cycle progression, as CDC6 and CDT1 depletion upon palbociclib release reduced chromatin-bound PCNA despite increasing levels of Cyclin A2 (Fig. 4e). Directly comparing the chromatin states after palbociclib release, with or without CDC6 and CDT1 degradation, revealed a scenario where i) ORC1 levels are elevated, suggestive of failed ORC exclusion, and ii) phosphorylation of MCM4 and MCM2 (Ser 53) is impaired, which are independent indicators of failed MCM double-hexamer formation (Fig. 4e). Degradation of CDC6 and CDT1 upon palbociclib release did not reduce the level of chromatin-bound CDC7 nor did it impair MCM2 phosphorylation on an CDC7-independent site (Ser27)56, suggesting that both the MCM substrate and CDC7 kinase are available, yet their functional interaction is blocked49,50,51,57. To test if these chromatin changes are not a general response to CDC6 or CDT1 degradation, we performed an analogous assay upon mimosine release, which revealed no difference in ORC1, PCNA or MCM phosphorylation status despite efficient CDC6 and CDT1 depletion (Supplementary Fig. 10c, d). Finally, replacing palbociclib with an independent CDK4/6 inhibitor, Trilaciclib, confirmed a significant reduction in DNA synthesis upon CDC6 and CDT1 depletion (Fig. 4f). Together, these data support a model where CDK4/6 activity controls a relevant pool of replication origins and acts prior to S-phase to generate productive MCM complexes on chromatin.
Fig. 4: CDK4/6-inhibition causes an origin licensing defect upstream of CDC6/CDT1 function, which can be sustained to trigger replication failure and aberrant mitosis in p53-deficient cells.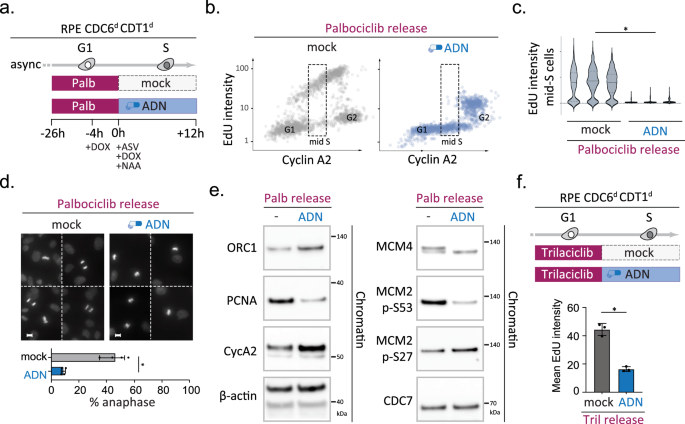
a–f Sequential CDK4/6 and licensing inhibition triggers mitosis with unreplicated DNA. a outline of the experiment. b QIBC analysis simultaneously monitoring DNA synthesis (EdU incorporation) and nuclear Cyclin A2 levels in single RPE CDC6d CDT1d cells 12 h after palbociclib release (>1000 cells per condition). Figure axes depict integrated fluorescence intensities (arb. units) per nucleus; Upon release cells were either mock treated (grey) and exposed to ADN treatment (blue); dotted line box highlights mid S-phase cells (classified based on CyclinA2). c violin plots depict distribution of EdU signals in three independent mid S-phase populations per condition, treated and classified as in (a, b) (n = 300 mid-S phase cells per replicate). Asterisk indicates a significant disparity between population means; n = 3 technical replicates per condition, p = 0.00002 (two-tailed paired t test). d mitotic phenotypes upon treatment described in (a); compound DAPI images show representative mitotic nuclei; scale bars, 10 μm; bar graph depicts quantification of the percentage of anaphase nuclei among mitotic cells; bars indicate mean % of anaphase-like nuclei; error bars indicate SD of four independent replicates. Asterisk indicates p = 0.0002 (two-tailed paired t test). e Immunoblots examining the chromatin occupancy of indicated proteins and/or phosphorylated epitopes upon treatment described in (a); the reduction of PCNA as well as MCM4 and MCM2 serine 53 phosphoryation confirm a licensing defect. β-actin served as loading control. Representative blots are shown, with the experiment repeated twice independently with consistent results. f Top panel depicts outline of the experiment and bar graph below shows mean integrated intensity of nuclear EdU signals for each condition (arb. units); error bars indicate SEM of three independent experiments; black dots indicate replicate means. Asterisk indicates a significant disparity between population means; p = 0.0004 (two-tailed paired t test). Source data are provided as a Source Data file.
CDK4/6 activity is required at the time of origin licensing
We next set out to establish when CDK4/6 activity is required during G1 phase and how relatively short pulses of CDK4/6 inhibition affect MCM loading and origin activity genome-wide. Previous live-cell imaging studies in CHO and MCF7 cells detected stable MCM loading throughout G1 phase but also found dynamic chromatin-MCM interactions to occur as early as telophase, implying that the first steps of origin licensing start already at mitotic exit58,59. When we exposed asynchronous RPE1 cells to a short 8-h pulse of Palbociclib, we detected a significant loss of MCM2 intensity in pre-extracted G1 phase nuclei (Supplementary Fig. 4e), indicating that non-transformed RPE1 cells require CDK4/6 activity during G1 phase or a few hours before (i.e. G2/M phase) for effective and stable MCM loading.
Under unchallenged conditions, the majority of G1 phase RPE1 cells are in a high MCM state while only a few are in a low MCM state, reflecting efficient MCM2 loading upon mitotic exit (Supplementary Fig. 11a, b). Upon a short 8-h pulse of Palbociclib, many cells remain stuck at the low MCM state and only a minority of RPE1 cells reached the high MCM state (Supplementary Fig. 11c). To study the role of CDK4/6 activity in G2/M versus G1 phase, we synchronised RPE1 cells by mitotic shake-off, either after a 3-h nocodazole pulse or a 3-h pulse with nocodazole and Palbociclib. Mitotic cells were released in the absence or presence of Palbociclib and 8 h later fixed and analysed for chromatin-bound MCM2 by QIBC (Supplementary Fig. 11d). Importantly, inhibiting CDK4/6 in G1 phase was sufficient to impair origin licensing Supplementary Fig. 11e). When CDK4/6 was inhibited also in G2/M, the origin licensing defect was enhanced, suggesting that licensing efficacy correlates to treatment duration and that a full block of MCM loading likely requires sustained CDK4/6 inhibition during mitotic exit and G1 phase.
To study how transient loss of CDK4/6 activity affects DNA replication initiation genome-wide, we made use of our time-resolved EdU-sequencing setup in which we allow RPE Cdc6d Cdt1d cells to progress through G1 phase in a synchronised fashion, block CDK4/6 activity either in early or late G1 phase and detect DNA replication in early S-phase cells (Fig. 5a). Flow cytometry analysis indicated that palbociclib treatment during early or late G1 phase is sufficient to cause a significant reduction of EdU-positive cells, confirming the need for sustained CDK4/6 activity to promote DNA replication (Fig. 5b). To define the interplay between CDK4/6 activity and the timing of origin licensing, we combined palbociclib treatments in early or late G1 phase with concurrent or alternating treatments with degron drugs (ASV, DOX, and NAA) and subsequently measured origin activity in early S phase (Supplementary Fig. 11). To directly compare the consequences of the different treatments, we determined the average EdU intensities of thousand top-ranked early S-phase origins for each condition (Fig. 5c). Focusing first on the palbociclib-only data, we found that CDK4/6 inhibition during early or late G1 phase diminished average origin activity by 65% or 50%, respectively. Notably, a 4-h palbociclib treatment during early G1 phase decreases origin activity more effectively than a 10-h treatment during late G1 phase, even when the former treatment allows more cells to enter S phase (Fig. 5b, c). Next, we examined how CDC6 and CDT1 degradation affected origin activity. In line with our previous observations, we find CDC6 and CDT1 to be needed in early and late G1 phase for efficient replication initiation (Fig. 5c, blue lines). We noted that CDC6 and CDT1 degradation until S phase caused average EdU peaks to be lower and wider than controls (Fig. 5c), which matches CDT1’s dual role in promoting origin licensing and limiting fork speed in early S phase, respectively60,61.
Fig. 5: CDK4/6 activity is needed at the time of origin licensing.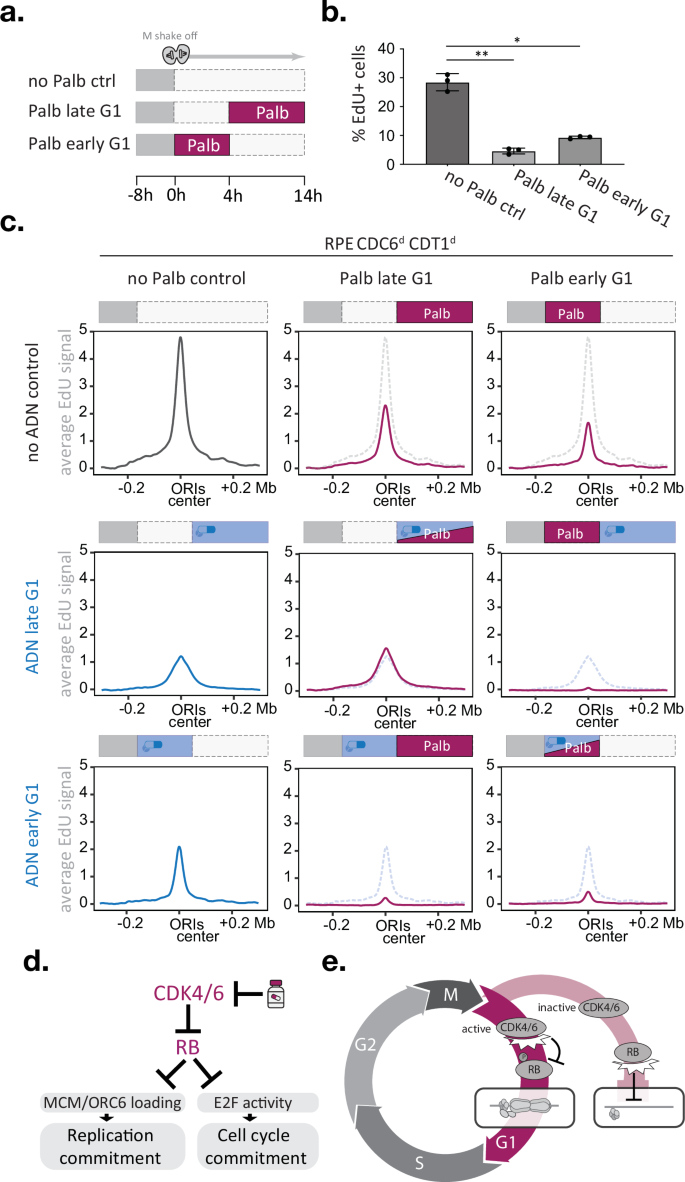
a–c Time-resolved EdUseq upon CDK4/6 inhibition and/or CDC6/CDT1 degradation in early or late G1 phase. a Outline of the palbociclib treatment timings. b bar graph depicts the percentage of EdU-positive cells by FACS analysis after the treatments indicated in (a); error bars indicate SD; black dots indicate replicate means. Statistical significance was determined using a two-tailed paired t test; n = 3) and asterisks indicate p < 0.05 (* p = 0.00038; ** p = 0.00019). c Average origin activity (i.e., mean σ values) at 1 Kb resolution around 1000 predefined, most active early S-phase origins after indicated palbocilib and/or ADN treatments. Schematic experimental outlines are depicted above each graph. Dotted lines indicate the average origin activity of the respective ‘no palbociclib’ control. d, e Proposed working models, d FDA-approved CDK4/6 inhibitors impede origin licensing and cell cycle commitment via RB regulation. e The CDK4/6-RB axis coordinates origin licensing and cell cycle commitment. In cells with high CDK4/6 activity, pocket proteins such as RB are inhibited by phosphorylation, which promotes both origin licensing and cell cycle commitment, thus ensuring efficient genome replication. In cells with low CDK4/6 activity, pocket proteins such as RB remain active, which stalls origin licensing and cell cycle commitment, thus preventing energy-consuming licensing activities in cells exiting the cell cycle. Source data are provided as a Source Data file.
Finally, we studied the effect of combining CDK4/6 and CDC6/CDT1 deficiency in a single G1 phase. Simultaneous palbociclib treatment and CDC6/CDT1 degradation during late G1 phase did not alter origin activity compared to CDC6/CDT1 degradation alone, suggesting that CDK4/6 activity acts in the same pathway as CDC6 and CDT1. Indeed, when CDC6/CDT1 degradation and CDK4/6 inhibition were implemented consecutively, i.e. switching from CDC6/CDT1 degradation during early G1 phase to CDK4/6 inhibition during late G1 phase, origin activity was diminished to 18% of mock-treated controls, which is two to threefold lower than either single treatment (Fig. 5c). The reciprocal experiment, switching from CDK4/6 inhibition during early G1 phase to CDC6/CDT1 degradation during late G1 phase, mirrors these results and reduced average origin activity to 17% of mock-treated controls. These findings demonstrate that efficient origin licensing requires CDK4/6 activity and the MCM loading machinery to be present at the same time. Blocking either of these activities during G1 phase is sufficient to reduce origin activity in early S phase. Protein degron approaches are ideally suited to study immediate effects upon protein loss, but for studying phenotype recovery these approaches rely on the target’s expression context. In line with CDK4/6 promoting CDT1 and CDC6 mRNA expression62,63, adding palbociclib during CDC6/CDT1 degradation in early G1 phase stalled the recovery of origin activity compared to CDC6/CDT1 degradation alone (Fig. 5c). Importantly, our biochemical data showed that palbociclib imposes licensing defects that are not restored by increased CDC6 and CDT1 levels (Supplementary Fig. 4d and Supplementary Fig. 7e) and involve ORC6 loading defects that manifest independently of CDC6 expression control (Fig. 3d and Supplementary Fig. 5c). The notion that short (4 h) palbociclib treatments in early G1 phase effectively diminished origin activity (Fig. 5c) supports a direct and upstream function for the CDK4/6 kinases in origin licensing.