
Human samples
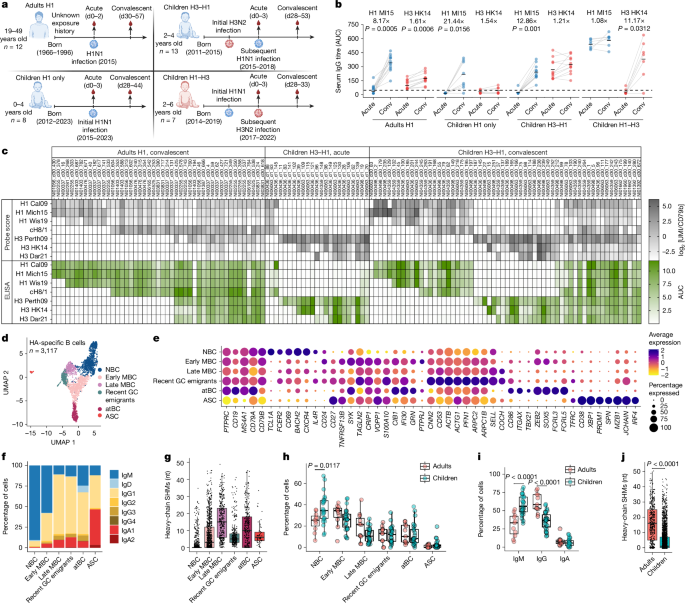
Serum and peripheral blood mononuclear cell (PBMC) samples were obtained from participants enrolled in two prospective community-based studies in Nicaragua: The Household Influenza Transmission Study (HITS) and the Nicaraguan Pediatric Influenza Cohort Study (NPICS). HITS is an intensive transmission study that enrols households after identification of a laboratory-confirmed influenza case and conducts intensive follow-up of household members. NPICS is an ongoing, longitudinal cohort of children aged 0–14 years, established to study the development of immunity to influenza from birth through annual serosurveys and active surveillance for acute respiratory illness. The sample sizes were not predetermined by statistical methods. These are observational studies, so no blinding or randomization was used.
PBMCs were isolated from venous blood by density-gradient centrifugation and cryopreserved for subsequent cellular immunology assays, as previously described41. NPICS samples were collected during the 2017–2018, 2018–2019, 2019–2020 and 2022–2023 seasons, whereas HITS samples were collected during the 2015–2016 season in Nicaragua, which typically spans June to December each year. These samples represent both the pre-infection (0–3 days between symptom onset) and post-infection (4–8 weeks after symptom onset) timepoints, enabling characterization of immune responses across the course of infection in a well-defined community setting. Written informed consent was obtained from parents or legal guardians of all participants, with assent from children as appropriate. Both studies received ethical approval from institutional review boards in Nicaragua and at the University of Michigan (HUM00091392 and HUM00088895).
Healthy infants for analysis of immune responses to primary influenza vaccination were recruited at paediatric primary clinics at Nationwide Children’s Hospital (NCH), Columbus, Ohio. Sera and PBMCs were collected after 1 month of vaccination as described above. The sample sizes were not predetermined by statistical methods, and no blinding or randomization was used. Written informed consent was obtained from parents or guardians before any of the study procedures. The study was approved by the NCH IRB (18-00591).
Cell lines and viruses
Madin–Darby canine kidney (MDCK) cells were purchased from ATCC and maintained in culture at 37 °C with 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM, Gibco) supplemented with 10% FBS (Gibco), 1% l-glutamine (Gibco) and 1% penicillin–streptomycin (final concentration, 100 units ml−1 penicillin, 100 μg ml−1 streptomycin, Gibco). Humanized MDCK (hCK) cells42 (in-house cell line generated by the Kawaoka laboratory) were maintained in culture at 37 °C with 5% CO2 in minimum essential medium (MEM, Gibco) containing 5% (v/v) newborn calf serum (Sigma-Aldrich), 0.225% sodium bicarbonate (Corning), 1× amino acids (Gibco), 1× vitamins (Gibco), 1× anti-anti (Gibco), 4 mM l-glutamine (Gibco), 2 μg ml−1 puromycin (InvivoGen) and 10 μg ml−1 blasticidin (InvivoGen). Expi293F suspension cells were purchased from Thermo Fisher Scientific and maintained in culture at 37 °C with 8% CO2 in Expi293F Expression Medium (Gibco) with shaking at 125 rpm. Cell lines were not authenticated or tested for mycoplasma at the lab level. Influenza viruses were grown in-house in MDCK cells or specific-pathogen free (SPF) eggs, collected, purified and titred (Supplementary Table 9).
Recombinant antigen and probe production
HA ectodomain sequences were synthesized by Integrated DNA Technologies (IDT) and cloned into a mammalian protein expression vector with a C-terminal foldon trimerization domain followed by AviTag and 6×His tag. Constructs were transfected using the ExpiFectamine 293 kit (Gibco) according to the manufacturer’s protocol. Supernatants were collected on day 5 after transfection and incubated with Ni2+-nitrilotriacetic acid (Ni-NTA) agarose (Qiagen). Agarose was then loaded onto a polypropylene gravity flow column (Thermo Fisher Scientific), washed with 20 mM imidazole in PBS and eluted with a solution of 500 mM imidazole, 20 mM Tris and 150 mM NaCl at pH 7.4. The eluate was buffer exchanged with PBS using a 100 kDa Amicon centrifugal column (Millipore). Purified HA proteins were stored at −80 °C (Supplementary Table 8).
For probes used in cell sorting, a Y98F mutation was introduced into all HA constructs, and constructs of the SARS-CoV-2 WT (D614G) receptor-binding domain (RBD) and chimeric HAs were generated as previously described43,44. All probes were synthesized, cloned, expressed and purified as described above. The purified probes were biotinylated (AviTag-specific) with BirA enzyme according to the manufacture’s protocol (Avidity). The unreacted biotin was removed by passage through a 7 kDa molecular mass cut-off desalting column (Zeba spin, Thermo Fisher Scientific). Biotinylated HAs were conjugated to TotalSeq-C PE streptavidin (PE-SA, BioLegend) and BV421 streptavidin (BV421-SA, BioLegend), whereas SARS-CoV-2 WT RBD was conjugated to APC streptavidin (APC-SA, BioLegend). Chimeric HAs (cH8/1 or cH4/3) and an empty control (no antigen, PBS only) were conjugated to TotalSeq-C non-fluorescent streptavidin (NF-SA, BioLegend). The amount of antigen needed for conjugation was calculated based on a 4:1 molar ratio of antigen to a fixed amount of 0.5 μg PE-SA, BV421-SA, APC-SA or NF-SA. Antigens were diluted in PBS to a final volume of 10 μl and SAs were added gradually to antigens five times on ice, 1 μl SA (0.1 mg ml−1 stock) every 10 min for a total of 5 μl (0.5 μg). The reaction was quenched with 5 μl 4 mM Pierce Biotin (Thermo Fisher Scientific) for 30 min for a total volume of 20 μl. Probes used for each sorting experiment were prepared on the same day.
Cell sorting for 10x Genomics single-cell sequencing
PBMCs were thawed and 0.5 million cells from each individual were aliquoted. The remaining PBMCs from each participant were pooled together for B cell enrichment using EasySep pan B cell magnetic enrichment kit (StemCell Technologies). Enriched B cells were stained with CD19 PE-Cy7 (BD Biosciences), TotalSeq-C CD79b (BioLegend) and antigen probes (HA-PE-SA, HA-BV421-SA, RBD-APC-SA, chimeric HA/empty-NF-SA) at 1:100 dilution in fluorescence-activated cell sorting (FACS) buffer (PBS supplemented with 2% FBS and 2 mM Pierce Biotin) on ice for 30 min. B cells were washed twice with FACS buffer and stained with 7-AAD viability staining solution (BioLegend). Populations that were 7-AAD−CD19+APC−PE+BV421+ were sorted as HA-specific B cells on the BD FACSMelody cell sorter (BD Biosciences). An aliquot of 0.5 million PBMCs from each individual was stained with CD19 PE-Cy7, CD4 BB515 (BD Biosciences) and a unique TotalSeq-C anti-human hashtag (BioLegend) at 1:100 dilution in FACS buffer on ice for 30 min. Aliquots were washed twice with FACS buffer and subsequently pooled together before staining with 7-AAD. Populations that were 7-AAD−CD19+ were sorted as CD19+ B cells. To account for low cell numbers, several thousands of CD4+ T cells from pooled PBMCs were sorted and mixed into HA-specific B cells as carrier cells. PBMCs from acute or convalescent timepoints were processed and sorted separately.
Sorted cells were immediately loaded onto the 10x Chromium X Controller to generate single-cell gel-beads in emulsion (GEM) using Chromium Next GEM Single Cell 5′ HT Reagent kits v2 (10X Genomics). 10x single-cell libraries of 5′ Gene Expression, V(D)J BCR and feature barcode (cell surface protein) were prepared according to the manufacturer’s user guide (CG000424 Rev B). Libraries were pooled and sequenced using NextSeq 1000/2000 P2 XLEAP-SBS Reagent Kit (300 cycles) on the NextSeq 1000 platform (Illumina) with the configuration of 26 cycles for read 1, 10 cycles for i7/i5 index and 150 cycles for read 2.
Computational analysis for single-cell sequencing data
Libraries of 5′ gene expression, V(D)J BCR and feature barcodes were demultiplexed using the cellranger mkfastq pipeline. FASTQ reads were mapped to the human genome (GRCh38-2020-A) either using a combination of cellranger_count and cellranger_vdj, or cellranger_multi (v.7.0.1 or newer). Downstream analyses were performed in R v.4.2.2 using Seurat (v.4.3.0 or newer), including quality control, data normalization, data scaling, dimension reduction (both linear and nonlinear), clustering, differential expression analysis, batch-effect correction, data visualization, and UMAP generation. Quality-control thresholds for each dataset were determined based on the distributions of detectable gene numbers (remove outliers with <200 or >3,000–5,000 based on dataset) and the percentage of mitochondrial genes (remove outliers with >5–10% based on the dataset). The donor identity of each cell was determined using an in-house hybrid demultiplexing approach that integrated results of cell hashing (hashtag) and a single-nucleotide-polymorphisms-based demultiplexing method (Souporcell) as previously described45. The following cells were filtered out: cells classified as doublets or unassigned based on hybrid demultiplexing; outliers in the distribution of detectable gene numbers or percentage of mitochondrial genes; and cells not expressing CD79A or expressing any of the non-B cell markers (LYZ, CD14, CD8A, GNLY, PPBP, CD3E, FCER1A, FCGR3A). Transcriptome data were normalized using a log-transform function with a scaling factor of 10,000, whereas cell surface protein data (TotalSeq antibody and antigen probe) were normalized using centred log-ratio normalization. Batch-effect correction analysis was performed using an Anchor method implemented in Seurat. After integration, we performed data scaling and linear dimension reduction using variable genes in principal component analysis and the top 15 principal components. High-quality cells were then clustered using the Louvain algorithm implemented in Seurat under a resolution of 0.6. Cells were clustered using the FindNeighbors and FindClusters functions. Further dimension reduction and visualization were performed using the RunUMAP function. DEGs for each cell cluster were identified using a Wilcoxon rank-sum test implemented in Seurat. B cell subset identity was assigned to each cluster based on DEGs and expression of stereotypical gene markers. We performed GSEA with DEGs from a putative recent GC emigrant cluster using the fgsea package v.1.24.0. Published gene sets were used for GSEA and relevant biological processes with adjusted P < 0.01 and normalized enrichment score > 2 were plotted (Extended Data Fig. 3h).
Single-cell BCR-sequencing analysis
Full-length V(D)J contigs were assembled with cellranger_vdj or cellranger_multi (v.7.0.1 or newer) and aligned to IMGT reference using IgBLAST v.1.20.0. The downstream analyses and annotations were done using an in-house single-cell multi-model analysis software VGenes (https://wilsonimmunologylab.github.io/VGenes/). Cells with ‘good BCR’ were filtered using the following criteria: cells passed the transcriptome quality control, V(D)J information is available for one heavy chain paired with one light chain, classified as full-length and productive by VGenes. Chord plots for B cell repertoires were generated in R using chordDiagram function of circlize package v.0.4.16.
Antigen specificity prediction through probe score
Unnormalized UMI counts were extracted from the Seurat ADT assay for cells with ‘good BCR’. A probe score for each antigen was calculated as the UMI ratio of antigen to CD79b and an empirical cut-off of 0.1 was set to distinguish positive from negative. UMI ratios shown in probe score heat maps were log2-transformed for better visualization. To account for the dilution effect in UMI per antigen for cells reactive to multiple antigens, cells were considered to be positive for a given subtype (H1 or H3) when the sums of probe scores of all HAs from the same subtype were higher than 0.1. Subtype reactivity of each cell was determined based on the following criteria: (1) cells positive for H1 were identified as H1 reactive; (2) cells positive for H3 were identified as H3 reactive; (3) cells with UMI > 0 for at least two antigens from each subtype and positive for both H1 and H3 were identified as H1/H3 cross-reactive; (4) cells positive for the empty probe (no antigen) were identified as polyreactive; (5) cells positive for neither H1 or H3 were identified as non-reactive. Cells belonging to the polyreactive, or nonreactive category, were considered non-specific and removed from the dataset of HA-specific B cells. Within H1-reactive cells, stalk binders were identified when scores of chimeric H1 (cH8/1) were higher than the mean scores plus 3 s.d. of all H1 probes, while head binders were identified otherwise. The predicted antigen specificities of subsets of B cells were validated with corresponding monoclonal antibodies by ELISA.
Serum ELISA
High-protein-binding microtitre plates (Costar) were coated with recombinant HA antigens at 2 μg ml−1 in PBS overnight at 4 °C. The plates were washed three times with PBS containing 0.05% Tween-20 (PBS-T) and incubated with 200 μl blocking buffer (PBS containing 0.1% Tween-20 and 3% skimmed milk powder) for 1 h at room temperature. Serums were threefold serially diluted in dilution buffer (PBS containing 0.1% Tween-20 and 1% skimmed milk powder) starting at 1:100. Plates were incubated with 100 μl serum dilutions for 2 h at room temperature. Horseradish peroxidase (HRP)-conjugated goat anti-human IgG secondary antibody (Sigma-Aldrich) was diluted at 1:3,000 in dilution buffer. The plates were incubated with 100 μl secondary antibody dilutions for 1 h at room temperature. Plates were washed three times with PBS-T and developed with 100 μl SigmaFast o-phenylenediamine dihydrochloride solution (Sigma-Aldrich) for 10 min in the dark. An additional 50 μl 3 M hydrochloric acid was added to stop the development reaction. Absorbance was measured at 490 nm on a microplate spectrophotometer (Bio-Rad). The area under the curve (AUC) for each serum sample was calculated in GraphPad Prism 10. The limit of detection was calculated as mean absorbance plus 3 s.d. of serum at the acute timepoint from H1-only children who were previously naive to influenza. All AUCs below the limit of detection were replaced by a fixed value of 15.
Recombinant monoclonal antibody and Fab production
Subsets of HA-specific B cell clones with ‘good BCR’ from different cohort groups or timepoints were randomly selected for monoclonal antibody expression. Notably, all H1/H3 cross-reactive clones were selected to enrich sample size for memory recall response. The immunoglobulin heavy and light chain sequences of these clones were synthesized as gene fragments by IDT or Twist Bioscience. To generate germline version of monoclonal antibodies, the variable regions of each clone were aligned to IMGT reference using IgBLAST v.1.20.0 and somatic mutations in heavy and light chains were both reverted. Unmapped nucleotides in CDR3 junctions were replaced with sequences of affinity-matured antibodies. Synthesized gene fragments were cloned into human IgG1, human κ or human λ expression vectors. The heavy and light chain constructs of each monoclonal antibody were transiently co-transfected into Expi293F cells using polyethylenimine (Polyscience). The supernatants containing secreted monoclonal antibodies were collected on day 5 and purified using protein A-agarose beads (Thermo Fisher Scientific) as described previously46. To generate Fab, the heavy-chain fragments of selected monoclonal antibodies were inserted into a modified human IgG1 vector with Fc constant region replaced by 6×His tag. The Fab version heavy chain and corresponding light-chain constructs were co-transfected and secreted Fabs within supernatants were purified according to the same protocol for purifying recombinant antigen, as described above, except that the buffer exchange was done using a 10 kDa Amicon centrifugal column.
Antigen-specific high-avidity ELISA
High-protein-binding microtitre plates were coated with recombinant monoclonal antibodies at 2 μg ml−1 in PBS overnight at 4 °C. Plates were washed three times with PBS-T and blocked with 150 μl PBS containing 20% FBS for 1 h at 37 °C. Antigen probes were generated as described before with unconjugated streptavidin (Invitrogen) and serially diluted 1:3 starting at 30 μg ml−1. The plates were incubated with 50 μl of probe dilutions for 1 h at 37 °C. HRP-conjugated rabbit anti-streptavidin polyclonal antibody was diluted 1:1,000 (Abcam) and 70 μl of antibody dilution was used to detect binding of probes. The plates were washed three times with PBS-T and subsequently developed with Super Aquablue substrate (eBiosciences). The absorbance was measured at 405 nm on a microplate spectrophotometer (Bio-Rad). Control antibodies with known binding characteristics were included on each plate and results were recorded when the absorbance of control reached 1.0 optical density (OD) units.
Competition ELISA
High-protein-binding microtitre plates were coated with 50 µl of A/Michigan/45/2015 HA at a concentration of 1 µg ml−1 and incubated overnight at 4 °C. Monoclonal antibodies with known epitope specificities were incubated at room temperature with EZ-Link Sulfo-NHS-Biotin (Thermo Fisher Scientific) for 4 h to be biotinylated and the unreacted biotin was removed by passage through a 7 kDa molecular mass cut-off desalting column (Zeba spin, Thermo Fisher Scientific). After blocking the plates with PBS containing 20% FBS for 1 h at 37 °C, 50 µl of each competing monoclonal antibody serially diluted at 1:2 ratio starting at 200 μg ml−1 was added in the coated wells for 2 h at room temperature. Then, 50 µl of each biotinylated monoclonal antibody was added at a concentration equal to twice its Kd and incubated in the wells with the competing monoclonal antibodies for 2 h at room temperature. After washing the plates three times with PBS-T, the wells were incubated with 100 µl of 1:1,000 diluted HRP-conjugated streptavidin (Southern Biotech) at 37 °C for 1 h. Super Aquablue ELISA substrate (eBiosciences) was then added, and the absorbance was measured at 405 nm on a microplate spectrophotometer (Bio-Rad). Biotinylated monoclonal antibodies were incubated in designated wells on each plate without any competing monoclonal antibody, and data were recorded when the absorbance of these wells reached an OD of 1–1.5 units. After subtracting the background, the percentage competition of each well was calculated by dividing the observed OD of that well by the OD reached by the positive control, subtracting this value from 1, and multiplying by 100.
Focus reduction neutralization test
The day before the experiment, 25,000 MDCK cells were added to each well of a 96-well plate. Serial threefold dilutions of monoclonal antibody in 50 μl MEM, starting at 200 μg ml−1, were mixed with an equal volume of 100 focus-forming units of virus for 1 h at 37 °C and added to MDCK cells. Overlay was prepared by mixing 50 μl 2% methylcellulose (Sigma-Aldrich) and an equal volume of monoclonal antibody dilutions in MEM supplemented with 1 μg ml−1 tosyl phenylalanyl chloromethyl ketone (TPCK)-treated trypsin. The virus–antibody mixture was removed after 1 h of incubation. The 100 μl overlay was added to cells and the plates were incubated for 20 h at 37 °C. Cells were washed with PBS, fixed with 100 μl 80% ice-cold acetone at −20 °C for at least 1 h and blocked for 30 min with 100 μl staining buffer (PBS containing 3% FBS). Cells were incubated with 100 μl mouse anti-NP antibody (clone A3, Millipore) diluted 1:1,000 in staining buffer, followed by 100 μl goat anti-mouse IgG HRP (Southern Biotech) diluted 1:1,000 in staining buffer for 1 h at room temperature. The plates were then washed three times with PBS and developed with 50 μl TrueBlue peroxidase substrate (SeraCare Life Sciences) in the dark for 5 min. Quantification of viral foci was performed using the S6 Universal M2 reader (ImmunoSpot). The inhibition percentage was calculated as: ((virus-only foci − sample foci)/virus only foci) × 100. The IC50 or FRNT50 for monoclonal antibodies was determined using the log[inhibitor] versus normalized response (variable slope) analysis in GraphPad Prism 10.
BLI analysis
MI15 H1, HK14 H3, NC99 H1, NC99 H1 N46D or MI15 H1 D46N were biotinylated and loaded at a concentration of 40 μg ml−1 onto a streptavidin biosensor (Forte Bio/Sartorius) for 300 s. After the sensor was equilibrated in kinetic buffer (PBS containing 0.02% Tween-20 and 0.1% bovine serum albumin) for 60 s, the sensor was first soaked into 500 nM Fab or IgG for 150 s as an association step and then soaked into kinetic buffer for 150 s as a dissociation step. The Kd of each Fab or IgG was calculated using Octet Analysis Software v.12.2.2.26 (Forte Bio/Sartorius). Results with coefficient of determination (R2) of <0.96 were removed and considered to be negative (no binding).
Generation of viral mutants
The HA sequences of five H1N1 A/Michigan/45/2015 mutants each with one major antigenic site (Sa, Sb, Ca1, Ca2, Cb) ablated were obtained from a previous study47. The influenza eight-plasmid reverse genetics system48 was used to construct the mutant viruses. In brief, DNA plasmids of HA mutants were cloned into a pHW2000 vector and transfected into cocultured MDCK-SIAT cells and human embryonic kidney 293T cells with the other seven influenza segments of PR8. After 72 h, the supernatants were collected. Viruses were plaque purified on MDCK-SIAT cells grown in DMEM (Gibco) containing 10% FBS (Gibco) and penicillin–streptomycin mix (Gibco). Individual plaques were picked and grown in fresh MDCK-SIAT cells. To confirm the HA sequences of the viral mutants, viral RNAs were extracted from the supernatant and HA segments were amplified and confirmed by Sanger sequencing.
HAI assays
A/Michigan/45/2015 H1N1 wild-type or mutant viruses were diluted to eight HA units per 50 μl. Then, 25 μl of the diluted virus was combined in duplicate wells with an equal volume of serially diluted monoclonal antibodies, starting from 100 μg ml−1 in PBS. The virus–antibody mixture was incubated for 30 min at room temperature. Next, 25 μl of 1% turkey red blood cells (Lampire) was added and incubated for 30 min at room temperature. The minimum effective concentrations were read based on the final dilution at which haemagglutination was observed. For the assay using A/Victoria/361/2011 H3N2 virus, the same protocol was followed using guinea pig red blood cells (Lampire) with monoclonal antibodies starting from 60 μg ml−1.
DMS
The HA1 mutant library was generated in the background of H1N1 A/Michigan/45/2015 (Genbank: MK622940.1, residues Leu87–Gly254) through saturation mutagenesis. The linearized vector was generated from a pCTcon2 yeast display plasmid by using 5′-GGCCGGCTGGGCCGCTGCTAAAACTGAAGCAATAACAGAA-3′ and 5′-GGCCTCGGGGGCCTGTACCCATACGACGTTCCAGACTACG-3′ as primers. Inserts were generated by two batches of PCRs, followed by overlapping PCRs. The first batch of PCRs consisted of 21 reactions, each with an equal molar mix of eight primers as the forward primer and a universal reverse primer 5′-GCCTTCGCCGGAGCCTGGCTTGC-3′. The forward primers for the first batch of PCRs are listed in Supplementary Table 10. These forward primers were named as cassetteX_N, in which X represents the cassette ID and N represents the primer number. Forward primers with the same cassette ID were mixed at equal molar ratio and used in the same PCR. The second batch of PCRs consisted of another 21 reactions, each with a universal forward primer 5′-ACCTCTATACTTTAACGTCAAGG-3′ and a unique reverse primer as listed in Supplementary Table 10. Subsequently, 21 overlapping PCRs were performed using the universal forward primer and the universal reverse primer. For each overlapping PCR, the template was a mixture of 10 ng each of the corresponding products from the first and second batches of PCRs. The complete insert was an equal molar mix of the products of these 21 overlapping PCRs. All PCRs were performed using PrimeSTAR Max polymerase (Takara Bio) according to the manufacturer’s instructions. PCR products were purified using the Monarch DNA Gel Extraction Kit (New England Biolabs).
Yeast cells were transformed by electroporation according to a previously described protocol49. In brief, 5 μg of the HA1 insert library and 4 μg of the corresponding purified linearized vector were added into 400 μl of conditioned yeast. The mixture was electroporated and transferred into yeast peptone dextrose medium supplemented with 4 ml of 1 M sorbitol and incubated at 30 °C with shaking at 225 rpm for 1 h. Cells were plated onto synthetic dextrose casamino acid (SD-CAA) plates and incubated at 30 °C for 40 h. Colonies were then collected in SD-CAA medium, centrifuged at 1,700 rcf for 5 min at room temperature and resuspended in SD-CAA medium with 15% (v/v) glycerol such that OD600 was 50. Glycerol stocks were stored at −80 °C until used.
A glycerol stock of the yeast display library was recovered in SD-CAA medium and induced in synthetic galactose raffinose casamino acid (SGR-CAA) medium. We initially used APC anti-HA.11 (16B12, BioLegend) at a final concentration of 1 μg ml−1 to sort the yeast cells based on expression level. After positive gating in the expression sort, PE-conjugated monoclonal antibodies were used at a final concentration of 10 μg ml−1 to gate the yeast cells for binding sort of HA1 variant library. Using the BD FACSMelody Cell Sorter (BD Biosciences), yeast cells were gated by no HA1 expression (PE−APC−), HA1 expression with no binding (APC+PE−) and HA1 expression with binding (PE+APC+). The sorted cells were recovered in SD-CAA medium. Frozen stocks were made and stored at −80 °C until used. FlowJo v.10.8 software (BD Life Sciences) was used to analyse FACS data.
Plasmids from the yeast cells were extracted using a Zymoprep Yeast Plasmid Miniprep II Kit (Zymo Research) according to the manufacturer’s protocol. The HA1 mutant library was amplified by PCR using forward recovered primers 5′-CACTCTTTCCCTACACGACGCTCTTCCGATCTTCCTGGGAAATCCAGAGTGTGA A-3′ and reverse recovered primers 5′-GACTGGAGTTCAGACGTGTGCTCTTCCGATCTCCAGTTGCTTCGAATGTTATTTT-3′. Subsequently, adapters containing sequencing barcodes were appended to the amplicon using primers 5′-AATGATACGGCGACCACCGAGATCTACACXXXXXXXXACACTCTTTCCCTACACGACGCT-3′ and 5′-CAAGCAGAAGACGGCATACGAGATXXXXXXXXGTGACTGGAGTTCAGACGTGTGCT-3′. Positions annotated by an X represented the nucleotides for the index sequence. All PCRs were performed using Q5 High-Fidelity DNA polymerase (NEB) according to the manufacturer’s instructions. PCR products were purified using PureLink PCR Purification Kit (Thermo Fisher Scientific). The final PCR products were submitted for next-generation sequencing using MiSeq v.3 PE300 (Illumina).
Next-generation sequencing data were obtained in FASTQ format. Forward and reverse reads of each pair-end read were merged by PEAR50. The merged reads were parsed by SeqIO module in BioPython51. Primer sequences were trimmed from the merged reads. Trimmed reads with lengths inconsistent with the expected length were discarded. The trimmed reads were then translated to amino acid sequences, with sequencing error correction performed at the same time as previously described52. The number of reads corresponding to each HA1 variant in each sample is counted. The frequency (F) of a mutant i at position s within bin n of replicate k was computed for each replicate as follows:
$${F}_{i,s,n,k}=\frac{\mathrm{read}\,{\mathrm{count}}_{i,s,n,k}+1}{\sum _{s}\sum _{i}(\mathrm{read}\,{\mathrm{count}}_{i,s,n,k}+1)}$$
A pseudocount of 1 was added to the read counts of each mutant to avoid division by zero in subsequent steps. We then calculated the HA1 expression ratio (ER) of a mutant i at position s of replicate k as follows:
$${\mathrm{ER}}_{i,s,k}=\frac{{F}_{\mathrm{PE}+(i,s,k)}}{{F}_{\mathrm{PE}+(i,s,k)}+{F}_{\mathrm{PE}-(i,s,k)}}$$
Subsequently, we calculated the escape score (ES) of each mutant i at position s of replicate k as follows:
$${\mathrm{ES}}_{i,s,k}=\frac{{F}_{\mathrm{APC}+(i,s,k)}}{{F}_{\mathrm{PE}+(i,s,k)}}\times {\mathrm{ER}}_{i,s,k}$$
The expression score of a mutant i at position s of was then calculated by taking the average of the expression scores between replicates.
In the HA1 yeast display library, structural destabilizing mutations could potentially impact the interaction between HA and all three tested antibodies. This would lead to high escape score for some mutations, despite not being part of the epitope53. Two antibodies, Hb-9 and Hb-5, have been used as control in the experiments. Notably, antibody Hb-5 binds to Sa only and Hb-9 does not bind to any of five immunodominant sites. Based on our mutant virus HAI experiments, we inferred that their binding epitopes differed from one another. Therefore, to mitigate the effects of these destabilizing mutations, we determined the final escape score (FES) of each mutant i at position s of replicate k by subtracting its average escape score against all five tested antibodies, as destabilizing mutations are more likely to exhibit high escape score across all five screens:
$${\mathrm{FES}}_{i,s}={\mathrm{ES}}_{i,s}-\frac{\begin{array}{c}{\mathrm{ES}}_{\mathrm{mAb}-129(i,s)}+{\mathrm{ES}}_{\mathrm{mAb}-235(i,s)}+{\mathrm{ES}}_{\mathrm{mAb}-687(i,s)}\\ \,{+\mathrm{ES}}_{\mathrm{mAb}-\mathrm{Hb}-5(i,s)}+{\mathrm{ES}}_{\mathrm{mAb}-\mathrm{Hb}-9(i,s)}\end{array}}{5}$$
To overlay the epitope of each antibody onto the HA structure, the average final escape score of position s was then calculated by taking the average of the final escape score of all mutants at that position:
$${\mathrm{FES}}_{s}=\frac{{\sum }_{i\in s}{\mathrm{FES}}_{i,s}}{|\{i\in s\}|}$$
The average final escape scores of each antibody were then projected onto the cryo-EM structure of A/Michigan/45/2015 HA (PDB: 6XGC).
Microneutralization
Twofold serial dilutions of the monoclonal antibodies (ranging from 480 μg ml−1 to 0.23 μg ml−1) were performed in MEM supplemented with 0.3% (v/v) bovine serum albumin and 0.00006% (v/v) TPCK. Each dilution was mixed with equal volume of 100 tissue culture infectious doses 50 (TCID50) of virus (so that the final monoclonal antibodies concentrations range from 240 μg ml−1 to 0.12 μg ml−1) and incubated at 37 °C for 1 h. The monoclonal antibody–virus mixtures were inoculated into MDCK cells (for avian viruses and H1N1 viruses) or hCK cells (for H3N2 viruses) in quadruplicates in a 96-well plate and incubated at 39 °C (for avian viruses) or 37 °C (For H1N1 and H3N2 viruses) with 5% CO2 for 5 days. The IC50 values were calculated according to the Reed–Muench method based on the observed cytopathic effect in the inoculated cells.
Mouse challenge experiments
All mice were housed (12 h–12 h light–dark cycle, 20–24 °C, 45–65% humidity) for at least 1 week before any experiments. 1 mg per kg of monoclonal antibody cocktails were intraperitoneally transferred into 6–8-week-old female BALB/c mice (Jackson Laboratories). 1 mg per kg of an irrelevant anti-RBD antibody (S728_1157) was also transferred into mice as an isotype (IgG) control to detect any non-specific protection. All antibodies were transferred 2 h before infection as a prophylactic treatment. Mice from each group (n = 5) were anaesthetized with a ketamine xylazine mixture and infected intranasally with 5 LD50 of mouse-adapted A/New Caledonia/20/1999 H1N1 virus, A/New York City/PV63249/2022 H1N1 virus or A/New York City/PV63311/2022 H3N2 virus in a total volume of 50 µl PBS. Mice were weighed daily for 14 days after infection; those losing 25% of their initial bodyweight were humanely euthanized. A sample size of five mice per condition was used based on a previously performed power analysis. Mouse challenge experiments were not blinded or randomized. All experiments were conducted according to protocols approved by the Icahn School of Medicine at Mount Sinai Institutional Animal Care and Use Committee (IACUC).
Cryo-EM sample preparation and data collection
Purified d30_103 and d30_240 Fabs were mixed with HK14 H3 HA and Mich15 H1 HA, respectively, at a molar ratio of 1:2 (HA protomer:Fab), and incubated at room temperature for 1 h before grid preparation. The final concentration of HA in each sample ranged from 0.7 to 0.8 mg ml−1. The samples were mixed with octyl-β-glucoside (OBG; final concentration 0.1%, w/v) to aid particle tumbling and applied to Quantifoil 1.2/1.3 300 mesh copper grids. Grids were plunge-frozen in liquid ethane using the Vitrobot Mark IV (Thermo Fisher Scientific) with a blot force of 1 and a blot time of 4–4.5 s at 100% humidity and 4 °C. Micrographs for the d30_103-HK14 H3 and d30_240-HK14 H3 complexes were collected on a Glacios 2 microscope (Thermo Fisher Scientific) operating at 200 kV with a Falcon 4i direct electron detector, while micrographs for the d30_103-Mich15 H1 and d30_240-Mich15 H1 complexes were collected on the Glacios microscope equipped with a Falcon IV detector. Automated data collection was carried out using EPU (Thermo Fisher Scientific) at a nominal magnification of 190,000, with a total exposure dose of around 45 e− Å−2. The pixel size was 0.718 Å for the HK14 H3 datasets and 0.725 Å for the Mich15 H1 datasets, with a nominal defocus range of −0.6 to −1.4 μm and −0.7 to −1.4 μm, respectively.
The purified H1N1 A/Solomon Islands/3/2006 HA protein was mixed with NI04359_d30_245 and NI01056_d30_604 Fab at 1:4 molar ratio (Supplementary Table 11) and incubated overnight at 4 °C before purifying by size-exclusion chromatography on the Superose 6 Increased 10/300 column (Cytiva) in 20 mM Tris-HCl pH 8.0 and 100 mM NaCl. The anchor Fab FISW8454 was also added into the complex of NI01056_d30_604 Fab and H1N1 A/Solomon Islands/3/2006 HA to reduce the preferred orientation of the complex. The complex peak was concentrated to around 3 mg ml−1 and mixed with 0.5% (w/v) n-octyl-β-d-glucoside (Anagrade) to a final concentration of 0.1% (w/v) just before loading onto the grid. Cryo-EM grids were prepared using a Vitrobot Mark IV machine. An aliquot of 3 μl sample was applied to a 300-mesh Quantifoil R1.2/1.3 Cu grid pretreated with glow-discharge. High-resolution cryo-EM videos were collected on an FEI Titan Krios at 300 kV with a Gatan K3 detector.
Cryo-EM data processing and model building
All datasets of d30_103 and d30_240 were processed using cryoSPARC (v.4.5)55. Dose-weighted video frame alignment was carried out using Patch motion correction in cryoSPARC live to account for stage drift and beam-induced motion. The contrast transfer function (CTF) was estimated using Patch CTF in cryoSPARC live. Micrographs were curated based on CTF fits; those worse than 7–8 Å were excluded due to poor quality. Individual particles were selected from an initial subset of several hundred micrographs using Blob picker. After several rounds of 2D classification, classes resembling the Fab–HA complex were used to pick templates for all micrographs. Clean particle stacks were selected through iterative rounds of 2D classification and subsequently used for ab initio reconstruction, followed by heterogeneous refinement to further remove junk particles. The resulting reference volume was used as an initial model for homogeneous and/or non-uniform refinement with C3 symmetry applied. Particles were subjected to 3D classification, and the best classes were further refined using non-uniform refinement with subsequent global CTF refinement, yielding the final map. For model building, the AlphaFold3-predicted model and the cryo-EM structure of Mich15 H1 (PDB: 7KNA) were used as the initial models for HK14 H3 and Mich15 H1, respectively. The antibody Fv models were generated using ABodyBuilder256. The models were fitted into the cryo-EM maps using UCSF ChimeraX57. The models were manually adjusted using Coot (v.0.9.8)58 and further refined through Rosetta Relax59 and real-space refinement in Phenix60. HAs were numbered according to the H3 numbering scheme, while the antibody Fv was numbered based on the Kabat numbering scheme. Buried surface area, epitope and paratope residues, and their interactions were analysed using the PISA server61 and Epitope Analyzer62. Buried surface area was calculated using a cut-off of >5 Å2, and hydrogen bonds were defined as atomic interactions with a distance of ≤3.5 Å. Structural figures were generated using UCSF ChimeraX57.
Datasets of NI04359_d30_245 and NI01056_d30_604 were processed with CryoSPARC55 (v.4.5). Videos were subjected to motion correction and CTF estimation, and particles were picked with CryoSPARC blob picker followed by 2D classification63. The best classes from blob picker were used as templates for CryoSPARC template pickers, and the resulting particles were cleaned up by multiple rounds of 2D classification before ab initio reconstruction. The best class from ab initio reconstruction was subjected to homogenous refinement, reference-based motion correction, another round of homogenous refinement, local and global CTF estimation, and non-uniform refinement. Antibody NI04359_d30_245 and NI01056_d30_604 complexes were processed with C1 symmetry. All maps were sharpened using DeepEMhancer64 and all of the initial atomic models were built using ModelAngelo65. The models were subjected to multiple rounds of manual refinement in Coot66 (v.0.9.8) and real-space refinement in Phenix60. This process was iterated for several cycles until no significant improvement in the model was observed. Validation statistics are listed in Supplementary Table 11.
HA sequence conservation analysis
Full-length human H3N2 and H1N1 HA protein sequences circulating from 1968 to 2024 and 1977 to 2024, respectively, were downloaded from the GISAID database. To minimize temporal sampling bias, up to ten sequences per year were selected. Multiple-sequence alignments were conducted using Clustal Omega, and the resulting alignments were used to generate sequence logos with WebLogo 3. The sequence logos were further manually refined and annotated in Adobe Illustrator.
All human H1 (1918–1957; 1977–2024) and H3 (1968–2024) protein sequences with complete HA2 regions were downloaded from NCBI and GISAID (as of 22 September 2024). Using a Python script, the natural occurrence frequencies of amino acid at position 46 of HA2 in each year were calculated for H1 and H3 separately. The results were plotted as bar graphs with the ggplot2 package in R (v.4.2).
Molecular dynamics simulation
Cryo-EM structures of d30_103 and d30_240 in complex with Mich15 H1 were used as the starting models for molecular dynamics (MD) simulations. Moreover, complexes of d30_103 and d30_240 with NC99 H1 were modelled based on the previously reported NC99 H1 structure (PDB 8TP5) and the Mich15 H1 complexes. The starting structures for MD simulations were prepared in the Molecular Operating Environment (Chemical Computing Group) using the Protonate3D tool67. These structures were then further processed using CHARMM-GUI68. Each model was placed into a cubic water box of TIP3P water molecules with a minimum wall distance of 12 Å from the protein, and the system was neutralized with K+ and Cl− ions to a final concentration of 0.15 mM69. For all simulations, parameters of the AMBER force field 19SB were used70. For the glycans, we used the GLYCAM force field, namely the GLYCAM-06j parameter set71. We then performed three repetitions of 1 µs of classical molecular dynamics simulations for each HA–antibody complex using Amber2472. MD simulations were performed in an NpT ensemble using pmemd.cuda73. Bonds involving hydrogen atoms were restrained by applying the SHAKE algorithm, allowing a time step of 2 fs (ref. 74). The Langevin thermostat was used to maintain the temperature during simulations at 300 K with a collision frequency of 2 ps−1, and a Monte Carlo barostat with one volume change attempt per 100 steps was applied75,76,77. The interaction energies were calculated with cpptraj using the interaction energy (LIE) tool78. The electrostatic interaction energies were calculated for all frames of each simulation and provided the simulation-averages of these interactions. To calculate the interactions and interaction frequencies of the binding interface, we used the GetContacts tool (Stanford University, https://getcontacts.github.io/). ChimeraX was used for visualization57.
Statistical analysis
All statistical analyses were performed using R v.4.2.2 or GraphPad Prism 10. Details of the statistics used in each experiment were indicated in the corresponding figure legends. P < 0.05 was considered to be significant, and P > 0.05 was considered to be not significant. The number of repeats for experiments and the specific tests for statistical significance used are indicated in the figure legends. Results without specifying the number of repeats were collected in one experiment. General data analysis and visualization were performed using Adobe Illustrator and Microsoft Word, PowerPoint and Excel.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.