
Extraction and purification of organic molecules from samples
The Ryugu A0480 (11.9 mg) and C0370 (8.3 mg) aggregate samples were allocated through the Third Announcement of Opportunity (AO3) by the Japan Aerospace Exploration Agency (JAXA) (Supplementary Fig. 1a). Further descriptions of the Ryugu samples are available in the Ryugu AO database (‘Data availability’). Microscope images and near-infrared reflectance spectra (2.0–4.0 μm) (Supplementary Fig. 1) were obtained using the MicrOmega hyperspectral microscope in the clean chamber of the JAXA curation facility before AO3 sample selection, which captured the freshest state of the materials14,15. These Ryugu aggregate samples are rich in organic matter14,15,17,21, and some relevant functional groups (–OH, –NH and –CH) were observed in a non-destructive spectroscopic analysis (Supplementary Data Fig. 1b,c). The data acquisition procedure is available elsewhere48. The CI1 Orgueil meteorite (30.6 mg; from the Natural History Museum of Denmark)21,26 was also analysed as a suitable reference due to its mineralogical and elemental similarities to the Ryugu samples29,30. Procedural blanks using baked sea sand (14.4 mg, FUJIFILM Wako Pure Chemical Corporation; 30–50 mesh) and baked serpentine (8.8 mg; 500 °C for 3 h) were prepared following the same protocol as the Ryugu samples and used to assess background signals.
All extraction and purification procedures were conducted on an ISO Class 5 clean bench within an ISO Class 6 clean room at Kyushu University. Each sample was suspended in ultrapure water and transferred to a 1.5 ml polytetrafluoroethylene (PTFE) tube using a glass Pasteur pipette. A total of 550 μl of ultrapure water was used to complete the transfer from the sapphire dish to the PTFE tube. Organic molecules were extracted at 25 °C with 15 min of ultrasonication. After centrifugation for 10 min at 10,000 rpm (equivalent to 14,093g), the supernatant was collected in a glass ampoule. The residue was rinsed with 200 μl of ultrapure water, and the rinse was combined with the supernatant to yield approximately ~640 μl in total, designated as the ‘H2O extract’. Altogether, 80% of the combined H2O extracts from the A0480 and C0370 samples were freeze-dried under reduced pressure for subsequent analysis.
The residues extracted from the Ryugu samples were suspended in 600 μl of 6 M HCl and then transferred to glass ampoules using glass Pasteur pipettes. After purging the headspace with dry N2 gas, the ampoules were flame-sealed and heated at 110 °C for 12 h. The HCl extraction procedure (temperature and duration) was identical to that previously applied to the Bennu sample27; however, in the present study, a preliminary water-based ultrasonic extraction at room temperature was performed to selectively isolate molecules that might otherwise be decomposed during acid treatment. Following the acid extraction, the supernatant and sample residues were transferred back into the same PTFE tubes used for the initial water extraction, using glass Pasteur pipettes. After centrifugation for 10 min at 10,000 rpm, the supernatant was collected into a glass ampoule. The residues were washed twice with 200 μl of ultrapure water, and the rinses were combined with the supernatant, designated as the ‘HCl extract’. Secondary minerals (for example, carbonates and phyllosilicates)22,29,30,49 also dissolved during the acid extraction and contributed to the composition of the HCl extract. The entirety of the combined HCl extracts from the A0480 and C0370 samples were freeze-dried under reduced pressure.
The freeze-dried H2O and HCl extracts were redissolved in 0.5 ml of 0.1 M HCl for desalting using an improved cation-exchange chromatography method10. A 0.5-ml aliquot of AG 50W-X8 cation-exchange resin (Bio-Rad Laboratories; analytical grade, 200–400 mesh, hydrogen form) was packed into a glass Pasteur pipette and preconditioned sequentially with 1.5 ml of 1 M HCl, ultrapure water, 1 M NaOH, ultrapure water, 1 M HCl and ultrapure water. The extract solution was then loaded onto the column. The resins were rinsed with 2.5 ml of ultrapure water to recover acidic, neutral and weakly basic compounds (designated as the ‘H2O fraction’). Subsequently, 2.5 ml of 10% NH4OH was applied to elute basic compounds, including most nucleobases (designated as the ‘NH4OH fraction’). As a result, four fractions (H2O–H2O, H2O–NH4OH, HCl–H2O, and HCl–NH4OH) were obtained from the Ryugu A0480 and C0370 samples. Despite the prewashing of the cation-exchange chromatography resin, trace amounts of N-containing molecules were detected in the NH4OH fractions of the procedural blank. These fractions were freeze-dried and reconstituted in 50–100 μl of ultrapure water for subsequent analyses. A fragment of the Orgueil CI meteorite (30.6 mg, purchased from a meteorite trading company) was extracted using the same protocol with ultrapure water and 6 M HCl. Note that the H2O extract of the Orgueil meteorite was not subjected to cation-exchange chromatography.
Carbon and nitrogen contents and the isotopic compositions of the extracted residues
The elemental abundances of carbon (C, wt%) and nitrogen (N, wt%) and their isotopic compositions (δ13C and δ15N, ‰ versus the international standards) in the residues extracted from the A0480, C0370 and the Orgueil meteorite were measured using an ultrasensitive nano-EA/IRMS system (Flash EA1112 elemental analyser/Conflo III interface/Delta Plus XP isotope-ratio mass spectrometer, Thermo Finnigan, Bremen)31,32. The masses of the analysed samples were 0.063 mg (A0480), 0.097 mg (C0370) and 0.125 mg (Orgueil). Each residue was loaded into precleaned smooth-wall Sn capsules (Lüdi Swiss AG), whose C and N blanks had been previously evaluated32. Isotopic compositions are expressed in conventional δ notation relative to the Vienna Peedee Belemnite for carbon and atmospheric air for nitrogen (denoted as ‘standard’), defined as:
$${\rm{\delta }}\equiv {10}^{3}({R}_{\mathrm{sample}}/{R}_{\mathrm{standard}}-1)\,({\permil }),$$
(1)
where R represents the 13C/12C or 15N/14N ratio of the sample (Rsample) or the standard (Rstandard). Calibration was performed using an international and three inter-laboratory standards—L-glutamic acid (USGS-41), L-tyrosine, L-alanine and nickel octaethylporphyrin—spanning δ13C values from −34.2‰ to +37.4‰ and δ15N values from +0.86‰ to +47.6‰. The isotope and elemental analyses were calibrated using standards spanning 1.5–27.1 µg (C) and 0.19–3.51 µg (N). The analytical uncertainties, determined from replicate analyses of the L-tyrosine standard, were ±0.27‰ (1σ, n = 8) for δ13C and ±0.50‰ (1σ, n = 9) for δ15N.
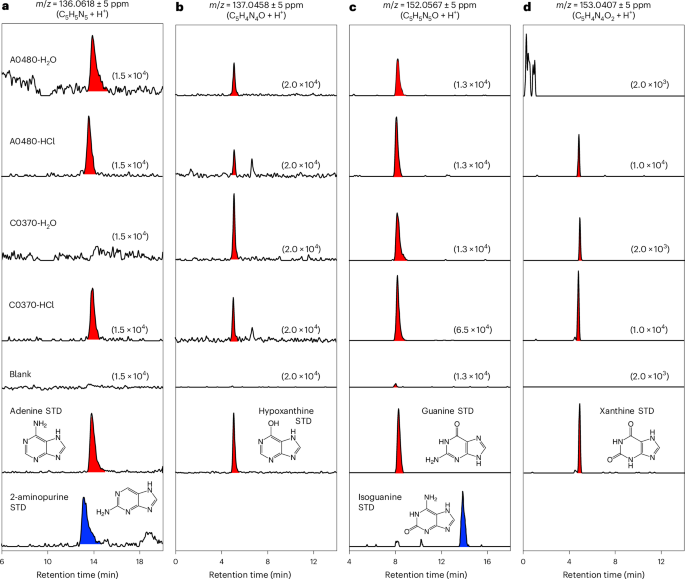
Analysis of nucleobases and other N-containing molecules
The H2O and HCl extracts derived from the Ryugu and Orgueil samples, alongside procedural blanks and authentic reference compounds, were analysed using a high-resolution online HPLC/ESI-HRMS system9,10,26,27. The instrumentation consisted of an UltiMate 3000 HPLC coupled to a mass spectrometer (Q Exactive Plus Hybrid Quadrupole-Orbitrap, Thermo Fisher Scientific), operated at a mass resolution of 140,000 at m/z = 200. A reversed-phase column was maintained at 40 °C for chromatographic separation.
Purine nucleobases were quantified using an isocratic elution programme with a pentafluorophenyl column (1.0 mm × 250 mm, 3-μm particle size; InertSustain, GL Sciences). The mobile phase consisted of 90% water (solvent A) and 10% acetonitrile containing 0.1% (by volume) formic acid (solvent B), delivered at a flow rate of 50 μl min−1 for 20 min. For the pyrimidine nucleobase analysis, a column (2.1 mm × 150 mm, 3-μm particle size; HyperCarb, Thermo Fisher Scientific) was used with a linear gradient of solvent A (water plus 0.1% formic acid) and solvent B (acetonitrile plus 0.1% formic acid), progressing from 99:1 at 0 min to 70:30 at 20 min, at a flow rate of 200 μl min−1.
The eluent was introduced into a HESI-II ion source (Thermo Fisher Scientific), operated at 280 °C for desolvation. The spray voltage and capillary temperature were set to 3.5 kV and 295 °C, respectively. Full-scan mass spectra were collected in positive ion mode across m/z ranges of 111–155 or 50–500 with mass accuracies better than 5 ppm. Mass calibration was occasionally performed using known ions, including protonated tyrosine (m/z = 182.08117), tert-butylamine (m/z = 74.09643) and its fragment ion (m/z = 57.06988), with an acetonitrile dimer (m/z = 83.06037) serving as the lock mass.
For confident identification of guanine and cytosine in the HCl–NH4OH fraction of the Ryugu C0370 sample, tandem mass spectrometry analyses were performed under the same ionization conditions as the full-scan analyses. The target positive ions were isolated using a quadrupole with a 0.4 atomic mass unit for the isolation window and fragmented by high-energy collisions with N2 gas. The resulting product ions were analysed using a mass spectrometer (Orbitrap) at a resolution of 140,000 at m/z = 200.
To cross-validate the detailed analysis of organic molecules in the Ryugu extracts, CE-HRMS was employed, as previously described21,26,50. Briefly, the CE-HRMS measurements were conducted with a capillary electrophoresis system (Agilent 7100, Agilent Technologies) coupled to a mass spectrometer (Q Exactive Plus, Thermo Fisher Scientific), an isocratic HPLC pump (Agilent 1260, Agilent Technologies), an adapter kit (G1603A CE-MS, Agilent Technologies) and a CE-ESI-MS sprayer kit (G1607A, Agilent Technologies). The CE and MS components were interfaced using a fused silica capillary (80 cm total length × 50 μm inner diameter), with a cation buffer solution (H3301-1001, HMT) serving as the electrophoretic electrolyte. Spectral data were acquired in positive ion mode over an m/z range of 60–900 at a resolution of 140,000 at m/z = 200.