
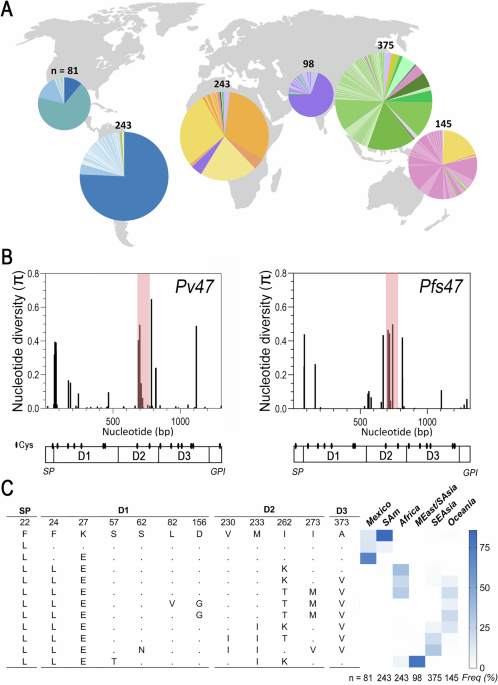
Worldwide genetic diversity and population structure of Pv47
The genetic diversity of Pv47 (PVX_083240; PVP01_1208000.1) was established by analyzing 1199 Pv47 gene coding sequences (Supplementary Data 1) obtained in 28 countries worldwide (Table S1). In total, 71 polymorphic sites were identified in the data set, most of which (68%) contained non-synonymous mutations (Table S2). A total of 209 Pv47 haplotypes were identified (Supplementary Data 2), the majority only present in a specific geographic area (Fig. 1A). Most of the Pv47 gene sequence is highly conserved, with amino acid sequence similarity between haplotypes ranging from 95% to 99.8%. However, there are clusters of non-synonymous polymorphisms close to the predicted N-terminus of domain 1 (Pv47-D1) and in the second domain (Pv47-D2), and one polymorphism is also frequently observed in the third domain (Pv47-D3) (Fig. 1B, left panel). Sites with nucleotide diversity (π) >0.05 were found to be non-synonymous substitutions (Fig. 1B).
Fig. 1: Geographic distribution of Pv47 haplotypes and genetic diversity of Pv47 and Pfs47.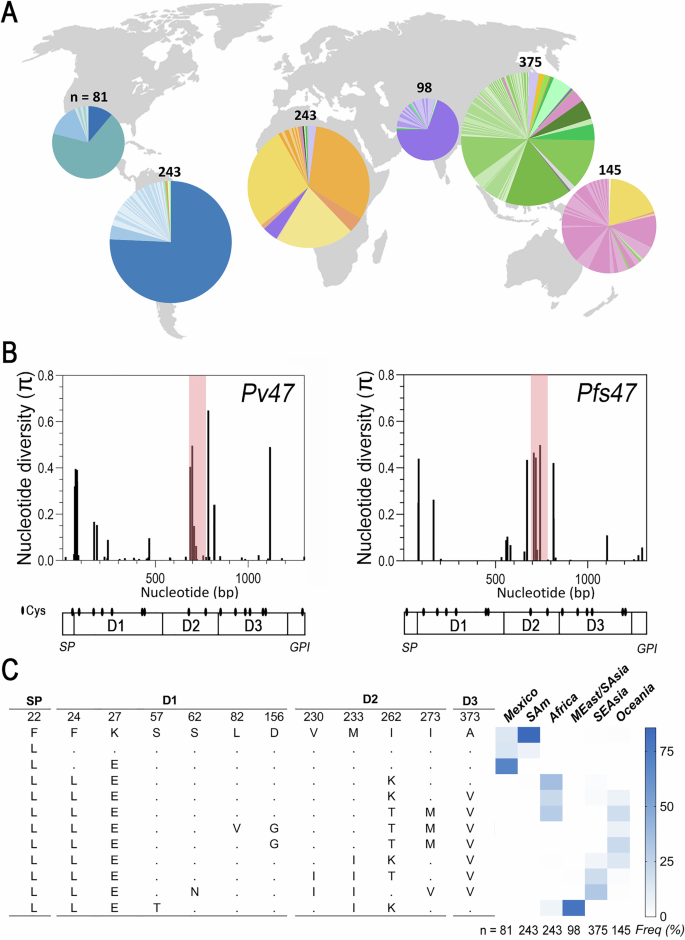
A Distribution of Pv47 protein haplotypes across different geographic regions. Haplotype frequency is represented by the size of the slice area in a pie chart. The color of each haplotype corresponds to the region where it is most frequent: South America is represented in blue; Mexico, Aqua; Africa, orange; Middle East/South Asia, purple; Southeast Asia, green; Oceania, pink. The number of Pv47 sequences analyzed per region are indicated. B Nucleotide diversity (π) per bp for Pv47 and Pfs47 gene coding sequences. The predicted protein domain structure and cysteine (Cys) locations (ovals) are indicated for both Pv47 and Pfs47. SP, signal peptide; D1, domain 1; D2, domain 2; D3, Domain 3; GPI, predicted glycosylphosphatidylinositol anchoring region. The region between two Cys in D2 is shaded in red. Source data are provided as a Source Data file 1. C Frequency of Pv47 aa haplotypes of polymorphisms with strong geographic structure. The Pv47 haplotypes shown are defined by SNPs that exhibit a marked population structure (FST > 0.2) between any of the regions compared and have a frequency greater than 5% in at least one of the geographic regions analyzed. SAm South America, MEast Middle East, SAsia South Asia, SEAsia Southeast Asia.
Pv47 presented high haplotype diversity (Hd) in the overall population (Hd = 0.95; Table S3). Interestingly, Pv47 sequences from East Asia/ Southeast Asia and Oceania had the highest haplotype diversity, Hd = 0.94 and Hd = 0.93, respectively, and the largest average genetic distance between sequence pairs (estimated as nucleotide diversity, π), with π = 0.0026 and π = 0.0035, respectively (Table S3). The rate of nonsynonymous polymorphisms per site was significantly higher than the rate of synonymous polymorphisms in the Pv47-D2, suggestive of natural selection, especially in South Asia, East Asia/Southeast Asia and Oceania’s populations (Table S3). At least three regions of Pv47, associated with non-synonymous polymorphisms in Pv47-D1, D2 and D3, presented Tajima’s D > 2, also suggestive of natural selection (Fig. S1A).
The frequency of the most common Pv47 protein haplotypes (frequency > 0.05) (Figs. 1C, S2) differed widely between geographic regions. Out of 71 polymorphic sites in the Pv47 coding region, 17 SNPs presented population structure with FST > 0.05, and 12 non-synonymous SNPs (causing residue changes F22L, F24L, K27E, S57T, S62N, L82V, D156G, V230I, M233I, I262K/T, I273M/V, A373V) had FST > 0.2 between some of the continental populations analyzed (Fig. 1C).
While most Pv47 sequences from South America (75.7%) were identical to the reference Pv-Sal I (from a strain collected in El Salvador), alternate Pv47 haplotypes F22L and K27E (Pv47-D1) were frequent (>0.7) in Mexico and other regions of the world. Alternate haplotypes F24L (Pv47-D1) and I262K/T (Pv47-D2) were also frequent beyond the Americas, while M233I (Pv47-D2) was common in South and Southeast Asia, and A373V (Pvs47-D3) was common in Africa, Southeast Asia, and Oceania. Allele encoding V230I and I273V (Pv47-D2) was frequent in Southeast Asia, while haplotype I273M (Pv47-D2) was frequent in Africa and Oceania (Figs. 1C, S2).
In general, haplotype network analysis shows separation of Pv47 haplotypes circulating in different continents (Fig. 2A), except for Africa, where the most frequent haplotypes are shared with Oceania or present at low frequency in Southeast Asia (Fig. 1C). A marked population structure was also obtained for P. falciparum Pfs47 (Table S5), consistent with previous reports9,18,19,20, which correlates with the different anopheline mosquito species that transmit malaria in a given region9.
Fig. 2: Genealogy and population structure of Pv47 haplotypes.
A Haplotype network (TCS) of the 209 Pv47 haplotypes identified world-wide. The geographic origin of each haplotype is indicated by a different color. The size of the circular node representing each haplotype is proportional to the number of samples with that sequence (a circle representing 10 samples is shown as reference). The perpendicular marks on the branches between haplotypes indicate the number of nucleotide substitutions separating the two haplotypes. NE Africa, Northeast Africa; America, Mexico and South America; MEast/S Asia, Middle East and South Asia; E Asia, East Asia; SE Asia, Southeast Asia. Source data are provided as a Source Data file 2. B Fixation index (FST) among Pv47 populations analyzed.
The largest genetic distances in Pv47 sequences were between populations from South America and those from Africa (FST 0.76–0.83), South Asia (FST 0.67–0.86), Southeast Asia (FST 0.63–0.86) and Oceania (FST 0.60–0.70) (Fig. 2B). There were also significant genetic differences between Middle East/South Asia and Southeast Asia (FST 0.25–0.66), and between Oceania and Middle East/South Asia (FST 0.39–0.50) and Southeast Asia (FST 0.28–0.42). Interestingly, significant genetic differences were also found within the New World, between South America and Mexico (FST 0.38–0.64) (Fig. 2B).
Worldwide genetic diversity and population structure of Pfs47
Detailed genetic diversity analysis of 4971 Pfs47 (PF3D7_1346800) sequences confirmed previous reports20,21 indicating that the most frequent non-synonymous polymorphisms in Pfs47 localize close to the predicted N-terminus of domain 1 (Pfs47-D1) and the second domain (Pfs47-D2), while a polymorphism in the third domain (Pfs47-D3) is less frequent (Fig. 1B, right panel). Pfs47 also presented high haplotype diversity in the overall population (Hd = 0.89; Table S4), with the highest haplotype diversity within South Asia (Hd = 0.87, Table S4). Notably, the average genetic distance between sequences from PNG was particularly large (π = 0.0022, Table S4).
The largest genetic differentiation in Pfs47 was between populations from South America and Asia (FST 0.79–0.94) followed by South America and Africa (FST 0.72–0.94) and Africa and Asia (FST 0.73–0.88) (Table S5). Significant population differences were also found within continents, for example, population differentiation of Pfs47 increased from West to East Africa and Madagascar (FST 0.32-0.38) (Table S5); while Central Africa (DRC) presented a modest difference with the rest of Africa (FST 0.09-0.25). In Asia, a significant population structure was observed between Southeast Asia and South Asia (Bangladesh)/Oceania (PNG) (FST 0.45–0.53), while in South America, Pfs47 presented marked population difference across the Andes, between the coastal regions of Colombia/Central America and the Amazonia in French Guyana/Peru/Brazil (FST 0.51–1.0; Table S5). Four regions of Pfs47, associated with non-synonymous polymorphisms in Pfs47-D1 and D2 presented Tajima’s D > 2, suggesting balancing selection, although other causes are possible (Fig. S1B).
Several Pfs47 amino acid polymorphisms differed between geographic regions. Pfs47 haplotypes private to South America differed from African haplotypes in non-synonymous polymorphisms in Pfs47-D2 (T236I, S242L, V247A, and I248L) which are nearly fixed between continents. These are polymorphisms that have been previously shown to be important for mosquito immune system evasion and parasite compatibility with anophelines9,10,11. Asian private Pfs47 haplotypes also differed from African haplotypes mostly in polymorphisms altering residues in Pfs47-D2 (I224N, T236I, L240I, I248L and N272Y) and one in Pfs47-D1 (L28I) (Fig. S3).
Within Africa, the frequency of the alternate Pfs47 haplotype E27D was high in East Africa, while in Central Africa haplotypes with E188D and N272Y/I were more frequent. In the New World, there was a major difference in Pfs47 populations between Colombia and Amazonia, with the non-reference haplotype I178V being fixed in coastal areas of Colombia, while the reference genotype encoding T68M was fixed in Amazonia. In Asia, there were also significant genetic differences in Pfs47 between Southeast Asia and Bangladesh, mostly due to differences in polymorphisms L28I, E55K, L240I and N272Y bp (Fig. S3).
Experimental evidence of selection of Pv47 by anopheline vectors
An. albimanus is the main vector of P. vivax in the lowlands of Chiapas, Mexico, while An. pseudopunctipennis is the main vector in the piedmont of the Sierra Madre mountain range14. Previous studies showed that the geographic distribution of three genetically distinct P. vivax populations correlates with the geographic distribution of these two vectors. Furthermore, side-by-side experimental infections in which both mosquito vectors were fed on blood from the same infected human host showed that genetically distinct P. vivax populations differed in their ability to infect these two mosquito vectors14,22. The authors suggested that the observed P. vivax population structure in Chiapas could be explained by differences in compatibility with these two vector species.
We investigated whether polymorphisms in Pv47 could explain the observed differences in vector compatibility by genotyping Pv47 in 43 isolates previously tested in experimental infections14 in which the infection prevalence was at least 2-fold higher in one of these two mosquito species and the difference in infection prevalence was statistically significant (Chi-square, p < 0.05) (Table S6). Of these P. vivax isolates, 15 had higher infections in An. albimanus, while 28 infected An. pseudopunctipennis more efficiently (Fig. 3A, Table 1, Table S6). A total of six Pv47 haplotypes were identified (Table 1). Three non-synonymous polymorphisms were present in single isolates (2%), while two of them F22L (88%) and K27E (65%), were frequent (Table 1). The K27E polymorphism in Pv47-D1, in proximity to the predicted N-terminal of Pv47, had a perfect correlation with the differences in vector compatibility. Those isolates with a positively charged lysine (K27) in this position had a significantly higher infection intensity in An. albimanus (Mann-Whitney; ****, P < 0.001), while those with a negatively charged glutamic acid (E27) had a significantly higher intensity of infection in An. pseudopunctipennis (Mann-Whitney; ****, P < 0.001), (Table 1, Fig. 3A).
Fig. 3: Association of the Pv47 haplotype polymorphism K27E with differential infectivity of two anopheline vectors.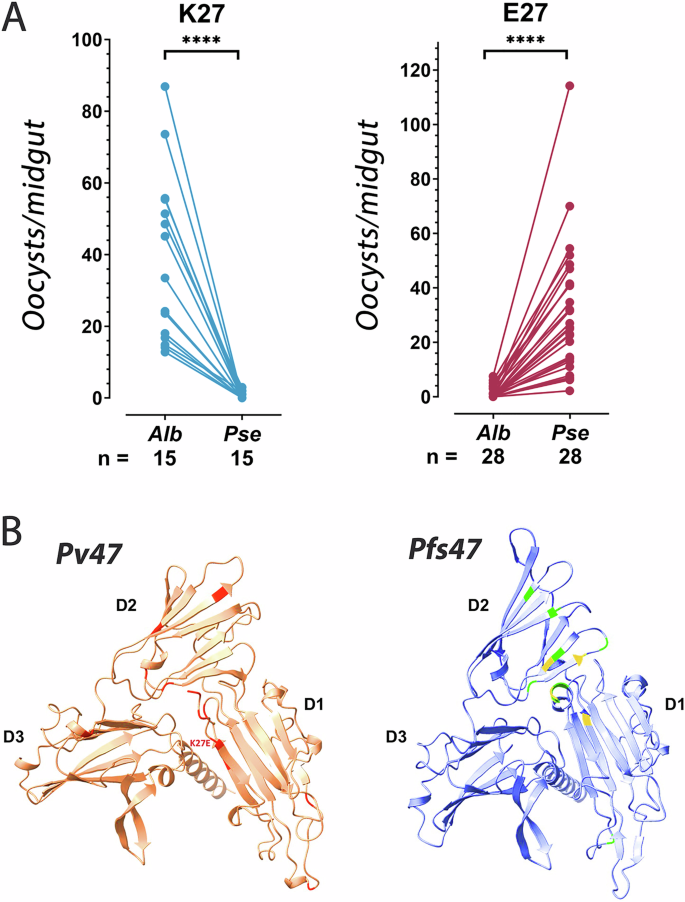
A Average number of P. vivax oocysts per midgut in Anopheles albimanus (Alb) and Anopheles pseudopunctipennis (Pse) mosquitoes infected with blood from the same individual infected with P. vivax parasites in the field that carry either the K27 or the E27 amino acid polymorphisms. The number of independent infections is indicated; two-tailed t-test; ****, p < 0.0001. Source data are provided as a Source Data file 1. B Protein structure of Pv47 predicted by Alphafold2 (left). The position of amino acid polymorphisms that present strong population structure (FST > 0.5; >0.05 frequency) are indicated in red color. Predicted location of amino acid polymorphism K27E is indicated. Protein structure of Pfs47 predicted by Alphafold2 (right). The position of amino acid polymorphisms that were previously shown to determine vector compatibility are indicated in light green and yellow. Predicted protein domains are indicated (D1, D2, D3).
Table 1 Pv47 haplotypes in Southern Mexico and their infectivity to Anopheles albimanus and Anopheles pseudopunctipennisPredicted molecular structure of Pv47 and Pfs47
In silico modeling of Pv47 and Pfs47 proteins using Alphafold223 predicted very similar structures despite having a modest level of amino acid identity (43%) (Fig. 3B). Both proteins consist of three 6-Cys s45/48 domains8 with a characteristic ß-sandwich fold formed by antiparallel and parallel ß-sheets (Fig. 3B). Domains D1 and D3 have the canonical 6-Cys pattern. In contrast, D2 is a shorter and degenerate s48/45 domain with only two cysteines, which is linked to the other two domains by flexible, less organized regions. Interestingly, the major polymorphisms in Pv47-D1, close to its N-terminal end, are predicted to be in spatial proximity to the protein surface where the major polymorphisms in Pv47-D2 are present. The major polymorphisms in Pfs47-D2 are known to be critical for immune evasion of the mosquito immune system and are major determinants of compatibility with different mosquito vector species through interaction with the Pfs47 receptor in the mosquito gut6,9,10. The predicted 3-D structure suggests that D1 amino acid polymorphisms in Pv47 -including K27E- may also interact with the mosquito Pfs47Rec to mediate immune evasion.