
Phylogenetic framework and HMMs of sulfur-cycling proteins
To discern sulfur-cycling genes or proteins from their functionally divergent homologues, phylogenetic analysis was conducted for 116 sulfur-cycling proteins (Supplementary Table 1). For each protein family, sequences of enzymes with biochemically validated functions, including related sequences of enzymes with divergent functions (outgroups), were identified by literature surveys and recovered from SwissProt61. Additional homologues of experimentally validated proteins in KEGG prokaryotic genomes were retrieved using KEGG BLAST Search (https://www.genome.jp/tools/blast/; E value: 10−4). Distant homologues that did not align properly with biochemically characterized proteins (alignment length covered <50% of query and target length) were removed. The resulting homologues were de-replicated using CD-HIT v4.8.1 (ref. 62), with longest sequences retained as representatives. For computational efficiency, different clustering identity thresholds (75–95%) were chosen for de-replication to ensure the total number of representative sequences for phylogenetic analysis did not exceed 500. The genome context of the representative KEGG homologues was analysed by retrieving genes located in a distance of fewer than n genes (n = 7–15), followed by annotation using biochemically characterized gene clusters based on BLAST analysis63. All representative KEGG homologues were further aligned with biochemically validated proteins and outgroups using Muscle v3.8.1551 (ref. 64). Poorly aligned regions were excised using TrimAl v1.4.rev15 (ref. 65). Protein phylogeny was inferred from the trimmed alignment using FastTree v2.1.7 (ref. 66) with -wag and -gamma options. Statistical support for each branch of the tree was estimated by nonparametric bootstrap (n = 100).
Information on reference sequences from biochemically verified proteins (for example, ingroup/outgroup, conserved residues or motif) and genomic contexts of all homologues were mapped on the tree. To identify monophyletic, orthologous clades within each tree, interior nodes of the annotated tree were scrutinized using the following criteria: (1) bootstrap support over 70%; (2) presence of at least one biochemically verified ingroup protein and absence of outgroup proteins; and (3) consistent gene neighbouring patterns and biochemical traits (thatis, catalytic residues and PFAM domain composition) among its members. All descendants of the identified clade were regarded as functional orthologs of the biochemically verified protein. If possible, existing definitions of orthologous clades from previous phylogenetic analysis of sulfur-cycling proteins was preserved, including the well-recognized clades in the phylogeny of DsrAB10 and Sqr67. For proteins for which the biochemically validated ingroup proteins formed polyphyletic groups, multiple monophyletic clades were proposed to fulfill our criteria.
To leverage our phylogenetic framework for large-scale homology searches, sequences from the defined monophyletic clades of sulfur-cycling proteins were used to build HMMs. A cut-off that optimizes the sensitivity and specificity of homology search was calculated for each HMM using receiver operating curve (ROC)68. This cut-off was embedded in the HMMER profile HMM file as the gathering threshold of the model (HMMER User’s Guide, p. 108; ref. 69). The performance of the newly developed HMMs was compared with that of six published sets of HMMs for sulfur metabolism genes, including those from KoFam70, TIGRFAM71, PFAM72, metabolicHMM73, DiSCo74, Teng et al.75 and HMS-S-S76. This was accomplished by querying each HMM against the phylogeny-curated protein dataset using hmmsearch in HMMER v3.2.1 with a predefined cut-off (http://hmmer.org/). The performance of the various HMM sets in detecting sulfur-cycling genes and proteins was assessed in terms of specificity, sensitivity, and F score (Supplementary Text). F score balancing both precision and recall of the homology detection was calculated using F score = 2 × (precision × recall) / (precision + recall).
Sulfur-cycling genes in bacterial and archaeal genomes
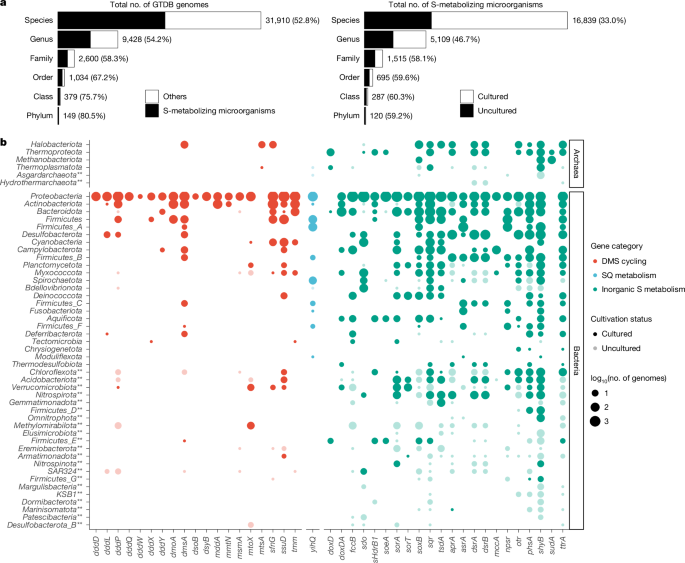
To provide a comprehensive overview of sulfur metabolism across bacteria and archaea, the phylogeny-derived HMMs were searched against all genomes in GTDB release 95 (ref. 23) using hmmsearch with the –cut_ga option. Each retrieved homologue was then searched against the full set of phylogeny-derived HMMs using hmmscan with –cut_ga, and annotated as the HMM showing the highest score. For initial screening, a subset of genes (n = 42) was selected as markers for specific sulfur metabolisms if the gene: (1) has been widely recognized as a marker for a specific sulfur metabolism, (2) encodes a catalytic subunit essential for the activity of enzymatic complex; or (3) on its own confers a specific sulfur redox transformation (see justification for each of selected genes in Supplementary Table 1). The retrieved homologues were further curated using our reference phylogeny of sulfur proteins. Specifically, the GTDB homologues were aligned with sequences contained in our reference phylogeny using Muscle. A maximum-likelihood tree was reconstructed from the alignment trimmed by TrimAl. The tree was overlaid with biochemical information and data on the genomic context of sulfur genes, and visualized using ggtree77. The physiological role of the GTDB homologues was interpreted on the basis of their evolutionary relationship with biochemically validated proteins and genome context. To predict the dissimilatory iron(iii) reduction potential, GTDB genomes were screened for marker genes involved in EET on the basis of homology search and/or motif analysis. Homologues of iron(iii) reduction genes with established HMMs in FeGenie database (https://github.com/Arkadiy-Garber/FeGenie/tree/master/hmms/iron/iron_reduction) were retrieved using hmmsearch from HMMER v3.2.1, with cut-off recommended by FeGenie (https://github.com/Arkadiy-Garber/FeGenie/blob/master/hmms/iron/HMM-bitcutoffs.txt). Additionally, homologues of MmcA gene, which is involved in dissimilatory iron(iii) reduction in Methanosarcina acetivorans78, were extracted using BLASTP on the basis of an e-value of 10−4. The outer membrane MHCs responsible for EET with metal oxides in anaerobic methanotrophs79 and putative electroactive bacteria80 were recognized on the basis of the following: (1) the presence of four or more haem-binding motifs (CXXCH); and (2) their predicted outer membrane or extracellular localization, as determined by DeepProLoc v1.0 (ref. 81).
Annotation and metabolic reconstruction of specific sulfur-cycling microbial lineages
The genomes of microbial lineages of interest were downloaded from the GTDB database. The protein-coding genes were predicted from the genome using Prodigal v2.6.3 with default setting. The predicted genes were annotated using KoFam70, PFAM72, and the EggNOG82 database. Additional metabolic pathways were predicted using HMMs (Supplementary Table 6) downloaded from dbCAN83, metabolicHMM73, CANT-HYD84, MicRhoDE85 and FeGenie86. For HMM-based annotation, the HMMs were used as queries to search against microbial genomes using hmmsearch from HMMER v3.2.1, with cut-off recommended by each database (-T, -domT or -cut_ga options). The cellular localization of the protein was predicted using Signalp v6.0 (ref. 87). The completeness of the KEGG metabolic pathway was calculated on the basis of the definition of each module. The KEGG module is defined with a logic expression of K numbers that records the composition of enzymes in the pathway. A particular metabolic module was considered to be present in the genome when: (1) the diagnostic/marker genes of the module were detected; and (2) the overall completeness of the pathway module was >70%. The environmental distribution of the GTDB species was retrieved by searching their GTDB species name in the Sandpiper database88. The occurrence of the GTDB species across biomes was downloaded as CSV from Sandpiper (https://sandpiper.qut.edu.au/) and further visualized with R v4.1.0.
Thermodynamic modelling
The Gibbs free energy associated with iron(iii)-dependent sulfur oxidation at environmentally relevant conditions was estimated by following the guidelines described previously89. In brief, the actual Gibbs free energy of reaction (ΔGr) was calculated using:
$$\Delta {G}_{{\rm{r}}}=\Delta {{G}_{{\rm{r}}}}^{0}+RT{\rm{ln}}{Q}_{{\rm{r}}}$$
where ΔGr0 refers to the standard Gibbs free energy of reaction, given in kJ mol−1; R and T are the universal gas constant (8.314 J K−1mol−1) and the temperature in Kelvin, respectively; and Qr is the reaction quotient. ΔGr0 values were calculated from the values of the standard Gibbs free energy of formation (ΔGf0) of reactants and products (Supplementary Table 7). Values of Qr were determined from the activity (ai) and the stoichiometric coefficient (vi) of the ith chemical species involved in the reaction using:
$${Q}_{r}=\prod {{a}_{i}}^{{v}_{i}}$$
The activity of the solvent (that is, pure water) and the solids (that is, ferrihydrite and FeS) were taken to be 1. The activity of dissolved ions was related to the concentration (Ci) using:
$${a}_{i}={\gamma }_{i}\times {C}_{i}/{{C}_{i}}^{0}$$
where γi denotes the activity coefficient; Ci0 represents the standard state concentration (usually 1 M). γi for cations (that is, Fe2+) and anions (that is, HS−, S2O32− and SO42−) in solutions of different ionic strength were retrieved from Amend et al.89. Sulfide speciation in aqueous phase across a range of pH was determined from the pH, and pKa1 (7.04) and pKa2 (11.96) of hydrogen sulfide.
Synthesis of ferrihydrite and poorly crystalline FeS
Synthetic ferrihydrite was prepared by titrating 1 M NaOH (Sigma Aldrich) into 0.1 M aqueous solution of FeCl3 6H2O (Carl Roth) under vigorous stirring until pH 7.5 was reached, as described90. The suspension was centrifuged (Centrifuge 5804 R, Eppendorf) at 4 °C, 12,857g and the ferrihydrite nanoparticles were washed thoroughly with deionized water to remove traces of chloride. The pellets were then freeze-dried (Alpha 1-4 LSCbasic, Christ) and stored at −20 °C for no longer than 3 weeks before use. The mineralogy was determined by LabRAM HR800 Raman microscope (Horiba Jobin-Yvon) equipped with a 532-nm neodymium-yttrium aluminium garnet laser and either 300 or 600 grooves/mm diffraction grating. Iron monosulfide (FeS; 30 mM) was prepared by mixing equal volume of 60 mM aqueous solution of Na2S.9H2O (Acros Organics) with 60 mM aqueous solution of FeCl2.4H2O (Sigma Aldrich) in an anaerobic chamber (Coy Lab) with 95% N2 and 5% H2 (O2 < 1 ppm) atmosphere. The initially precipitated FeS is often designated as ‘amorphous FeS’ or ‘poorly crystalline FeS’91. The dissolved sulfide in the FeS stock is less than 50 µM. The FeS solution was freshly prepared and used on the same day.
Cultivation of D. alkaliphilus DSM 19089
D. alkaliphilus (DSM 19089, ATH2) was purchased from the German Collection of Microorganisms and Cell Cultures GmbH (DSMZ). The bacterium was cultivated at room temperature in an alkaline mineral medium (pH 9.3) containing 3 g NaCl (Carl Roth), 0.25 g K2HPO4 (Merck), 6.5 g Na2CO3 (Carl Roth), and 15 g NaHCO3 (Sigma Aldrich) per liter of medium. After autoclaving, the medium was cooled down under N2 atmosphere and supplemented aseptically with 1 ml liter−1 of following components (all stored under anoxic conditions): 4 M NH4Cl (Sigma Aldrich), 1 M MgCl2 (Sigma Aldrich), trace element solution, Se-W solution, and four different vitamin solutions (DSMZ medium 1104). The culture was routinely grown under nitrate-reducing, sulfide-oxidizing conditions in 500 ml Schott bottles27, with 2 mM Na2S 9H2O and 1.2 mM KNO3 (Sigma Aldrich). This yielded a culture with an optical density at 600 nm (OD600) of ~0.040, corresponding to a cell density of ~1.3 × 108 cells per ml. To test alternative growth modes, five incubation experiments were conducted, each supplemented with different electron donors and acceptors (details provided below). For all experiments, regularly maintained cultures (30 ml) that have been depleted in sulfide (< 100 µM) and nitrate (< 10 µM) were used as inoculum. Incubations were set up in 60 ml serum bottles and sealed with butyl rubber stoppers in the anaerobic chamber (N2:H2 = 95:5). Each culture was then flushed with pure N2 to remove H2 in the headspace, and incubated in the dark at room temperature. All incubations, abiotic and biotic controls from each experiment were set up in triplicates.
(1)
Incubations with sulfide and nitrate. The incubations were set up by supplying 2 mM sulfide and 2 mM nitrate to 30 ml pre-growns cells in 60 ml serum bottles. Sulfide and nitrate was spiked using syringes flushed with pure N2. The growth was monitored by phase-contrast microscopy and by the measurement of sulfide and sulfate over 3 days.
(2)
Incubations with elemental sulfur. The incubations were initiated by adding 0.1 g elemental sulfur in 3 ml MilliQ water (Sigma Aldrich) to each of the serum bottles, followed by autoclaving at 110 °C for 60 min. After sterilization, 30 ml pre-grown cells were inoculated into the S(0) suspension (approximately 94 mM) and incubated under an N2 atmosphere for 15 days. Microbial activity was monitored by measuring sulfide and/or sulfate.
(3)
Incubations with ferrihydrite and formate. Synthetic ferrihydrite (0.2 g) was ground into fine particles with an agate mortar and pestle before being added to the culture. Assuming ferrihydrite has a composition92 Fe(OH)3, the final concentration of Fe(iii) was approximately 62 mM. Formate was spiked anoxically using a syringe to a final concentration of 10 mM. To test the coupling of ferrihydrite reduction and formate oxidation, parallel cultures were set up with either ferrihydrite or formate alone. The culture activity was monitored by measuring total Fe(ii) and formate concentrations over a 15 day incubation.
(4)
Incubations with ferrihydrite and poorly crystalline FeS. Synthetic ferrihydrite (0.2 g) was supplied to the cultures as described in the incubation (3). To amend poorly crystalline FeS, 1 ml stock solution of freshly prepared FeS (30 mM) was anoxically spiked to the cultures using syringes, resulting in a final FeS concentration of 1 mM. Abiotic controls were prepared using 30 ml autoclaved cells as inoculum to test for chemical reactions between ferrihydrite and poorly crystalline FeS. Biotic controls amended with either ferrihydrite or FeS were set up to assess the impacts of residual sulfide and/or nitrate on culture activity. Cultures were sampled daily over 5 days for sulfate and total Fe(ii) measurements.
(5)
Incubations with ferrihydrite and dissolved sulfide. The cultures were prepared similarly as incubation (4), but with dissolved sulfide replacing FeS. Due to the rapid chemical reaction between dissolved sulfide and ferrihydrite, dissolved sulfide was anoxically spiked daily at a concentration of 1 mM using N2-flushed syringes. Abiotic controls and the sulfide-only biotic controls received dissolved sulfide at the same concentration and frequency. To trace the transformation of S and Fe over 5 days, subsamples were taken daily for measurement of S(0), total Fe(ii), sulfate, and Cline-extractable sulfide before the addition of sulfide. The consumption of dissolved sulfide in the cultures was monitored by sampling at 2, 10, 20, 35, 50 and 70 min after the spike of sulfide. The kinetics of sulfide consumption were modelled as a first-order reaction. The rate constant was estimated using the exponential decay model in the drm() function from the drc R package93. To compare sulfate formation patterns with and without sulfide, cultures incubated with ferrihydrite and sulfide were sampled for sulfate measurement following two phases after the 1st sulfide spike. During phase I, detectable sulfide was present in the culture, and the samples were collected at 0, 11, 21, 37, 53 and 70 min of the incubation. Phase II, spanning the next 23 h, began once sulfide was depleted, with samples taken at 3, 5.5, 8.33, 12.33, 20.25 and 24 h. As a control for phase II, cells were incubated with chemically sulfidized ferrihydrite. Specifically, 1 mM sulfide was firstly added to 0.2 g ferrihydrite (approximately 62 mM) with 30 ml autoclaved cultures for chemical reaction. After 70 min, the reaction mixture was centrifuged (12,857g; room temperature) under anoxic conditions, and 30 ml of active cells were inoculated to resuspend the solid phase compounds (for example, FeS and S(0)) produced by chemical reaction between sulfide and ferrihydrite. Samples were collected from cultures for sulfate measurement at the same time intervals as those in phases I and II.
To test whether the microbial process can outperform the chemical process in transforming sulfide with ferrihydrite, the incubation (5) was repeated using ca. 50 µM sulfide instead of 1 mM. In this experiment, a small amount of sulfide was spiked three times at 1.5-h intervals into ferrihydrite-amended cultures, abiotic controls, and sulfide-only biotic controls. After each spike, subcultures (~ 0.3 ml) were collected at 2, 5, 10, 15, 20, and 25 min for dissolved sulfide measurements. Two biological replicates were performed for each treatment. To verify the reproducibility of the observed sulfide consumption pattern, incubations were conducted using inocula at different cell densities (OD600 of 0.042, 0.075, and 0.086). To quantify the transformation of spiked sulfide during the incubation, independent cultures were set up using an inoculum with an OD600 of 0.072 and supplied with eight spikes of sulfide. Ferrihydrite-amended cultures, abiotic controls, and sulfide-only controls received ca. 50 µM sulfide at 1.5-h intervals, whereas ferrihydrite-only biotic controls were spiked with anoxic water. Three replicate incubations were performed for each treatment. Subsamples were taken every three hours for concentration measurement of S(0), total Fe(ii), sulfate and Cline-extractable sulfide.
Chemical analysis of metabolites
To monitor the dynamics of metabolites in the incubation experiments, subsamples of the culture were taken periodically with sterile syringes flushed with pure N2 as described above. HCl-extractable Fe(ii) was determined by adding 0.1 ml sample aliquots to 0.2 ml 0.75 N HCl. The sample was immediately centrifuged for 15 min at 12,044g. Fe(ii) in the resulting 0.5 N HCl was measured using the ferrozine assay. Previous studies have shown the 0.5 N HCl treatment allowed quantitative extraction of the solid phase Fe(ii) associated with the surface of iron oxides, Fe(ii) from FeS, and the dissolved Fe(ii) in the Fe/S system7,94. Therefore, we referred to HCl-extractable Fe(ii) as total Fe(ii).
Aqueous and total sulfide were determined using spectrophotometric methods. To measure dissolved sulfide, approximately 0.3 ml subculture was filtered through a 0.2 µm membrane (CHROMAFIL). The dissolved sulfide in the filtrate (0.1 ml) was fixed by 0.25 ml 3% w/v zinc acetate dihydrate (Sigma Aldrich), followed by quantification using the Cline method95. The filtered sample from the incubation with ferrihydrite and 1 mM dissolved sulfide showed black colour, indicating the formation of FeS particles smaller than 0.2 µm. The sulfide associated with this FeS fractionation was approximated as HCl-extractable Fe(ii), assuming a 1:1 stoichiometry. The total sulfide was determined as Cline-extractable sulfide. The Cline reagent contains 6 N HCl that dissolves some solid sulfides (for example, freshly formed FeS), and thus the Cline-extractable sulfide comprises dissolved sulfide and HCl-reactive solid phase sulfide. Total sulfide in the Fe/S system is typically determined as acid volatile sulfide. Acid volatile sulfide was not analysed here owing to the large uncertainties inherent to this methodology91,96.
Sulfate and formate concentrations in the incubations were determined by capillary electrophoresis techniques. Sample preparation for sulfate measurement involved fixation of 100 µl subsample with 10 µl 3% w/v zinc acetate, dilution with 890 ul MilliQ water, filtration through a 0.2 µm membrane, and addition of 1 mM chlorate as the internal standard. The standards were prepared by adding defined amounts of sulfate (Sigma Aldrich) to the alkaline medium, followed by the same treatment procedure as described for samples. The sulfate content in the prepared samples/standards was measured using an Agilent 7100 capillary electrophoresis system (Agilent Technologies), equipped with a capillary (72 cm × 72 µm internal diameter; Agilent Technologies) and a diode array UV-vis detector (DAD). Electrolytes for anion separation contains 2.25 mM pyromellitic acid (Sigma Aldrich), 1.6 mM triethanolamine (Sigma Aldrich), 0.75 mM hexamethonium hydroxide (Sigma Aldrich), and 6.5 mM NaOH97 at pH 7.8 ± 0.1. Anion separation was implemented at a voltage of −30 kV. The data were acquired through indirect UV detection at a wavelength of 350 nm with a bandwidth of 60 nm, and a reference wavelength of 245 nm with a bandwidth of 10 nm. For the formate measurement, 900 µl MilliQ water was added to 100 µl samples/standards (Sigma Aldrich), which were then filtered through a 0.2 µm membrane. l-malate (Sigma Aldrich) was added to the filtrate as the internal standard. Organic Acids Buffer for capillary electrophoresis (pH 5.6; Agilent Technologies) was used as electrolytes, and the separation conditions, including DAD and capillary electrophoresis settings, were configured according to manufacturer’s instructions. All electropherogram data were analysed with the Agilent ChemStation.
Elemental sulfur was measured using high performance liquid chromatography (HPLC). One-hundred microlitres of sample was fixed with 10 µl of 3% w/v zinc acetate. Then, 300 µl chloroform was added, and the mixture was shaken at 500 rpm for 1 h. The elemental sulfur in chloroform phase was then measured using a Dionex UltiMate 3000 UPLC system, equipped with an UltiMate 3000 pump (0.2 ml min−1), a column Compartment (25 °C), a column Waters ACCQ-TAG ULTRA C18 1.7 µm × 2.1 × 100 mm, and an UltiMate 3000 Variable Wavelength Detector (UV) (wavelength 254 nm). The isocratic elution with 100% methanol was applied. With these adjustments, the peak appeared after 3.4 min. Data were analysed with Dionex Chromeleon software.
Microscopy of D. alkaliphilus incubated with ferrihydrite and sulfide
For scanning electron microscopy (SEM), transmission electron microscopy (TEM) and fluorescence microscopy, cultures incubated with ferrihydrite and sulfide (daily spike of 1 mM) for 5 days were fixed in 2% glutaraldehyde or 2.3% formaldehyde, respectively. For SEM imaging, solid iron phase iron was allowed to settle without centrifugation, carefully washed with MilliQ water, and transferred to 100% ethanol. Samples were then dried using rapid chemical drying with hexamethyldisilazane and mounted on aluminium stubs with double-sided sticky carbon tape and sputtered with Gold (JEOL JFC-2300HR). The images were taken with a Scanning Electron Microscope (JEOL IT 300 LAB6EOL) with Secondary Electron Detector (SED) and Backscattered Electron Detector (BED-C) at 20 kV.
For TEM imaging, cultures were treated with a solution containing 50 g l−1 sodium dithionite, 0.2 M sodium citrate and 0.35 M acetic acid (hereafter termed dithionite solution) as previously described98. After dissolution of solid iron phase, cells were pelleted at low speed (2,300g) to minimize shear forces and washed with MilliQ water before suspending cells in MilliQ water. For negative staining, 4 µl of sample was incubated for 1 min on a formvar-filmed and carbon-coated grid (200 mesh, Cu) and excess liquid was removed with a filter paper. A drop of stain (2.5% gadolinium acetate) was applied and immediately removed. Samples were examined in a TEM EM 900 N (Zeiss) at 80 kV.
For fluorescence microscopy, the formaldehyde-fixed cultures were resuspended and a subsample was filtered onto a 0.2 µm pore size polycarbonate membrane (Millipore). Cells on the filter were stained with a 1× SYBR Green solution, and images were acquired using a epifluorescence microscope (Zeiss Axio Imager M1 with an AxioCam MRm).
Monitoring the growth of D. alkaliphilus during incubation experiments
Growth was monitored by cell counting for cultures incubated under 4 different conditions: (1) ferrihydrite (approximately 62 mM Fe equivalent) and periodic spike of approximately 50 µM dissolved sulfide (sulfide was spiked 40 times over 5 days, with one spike every hour and 8 times per day); (2) ferrihydrite (approximately 62 mM Fe equivalent) and daily spike of 1 mM sulfide; (3) ferrihydrite (approximately 62 mM Fe equivalent) and periodic spike of FeS (approximately 1 mM Fe equivalent); and (4) nitrate (4 mM) and 2 spikes of sulfide at concentration of 1–2 mM. The setup of the cultures and controls was the same as described in ‘Cultivation of alkaliphilus DSM 19089’ except that a lower starting cell density (3–5 × 107 cells per ml) was used. During each of the incubation experiments, subcultures (450 µl) were sampled periodically and preserved in 2.3% formaldehyde (final concentration). Before counting, 500 µl dithionite solution was added to 50–100 µl of fixed cells to dissolve the FeS and ferrihydrite particles. After dissolution of solid iron phase (within 10–15 min), 100 µl of each sample was diluted in 900 µl of 1× phosphate-buffered saline (PBS). The suspension was then sonicated using a SONOPULS ultrasonic homogenizer (Bandelin, Berlin, Germany) at 25% power with a cycle setting of 2 for a total of 30 s. Cells were subsequently stained with SYBR Green 1× (ThermoFisher) and incubated for 10 min at room temperature in the dark. Flow cytometric analysis was performed using a CytoFLEX S flow cytometer (Beckman Coulter) equipped with a blue 488 nm laser. SYBR Green fluorescence was detected using a 525/40 nm bandpass filter. A fluorescence threshold was applied on the SYBR Green signal to exclude background events. For each sample, 80–100 µl was measured. Data were gated on SYBR Green–positive cells displaying fluorescence shifts relative to unstained controls to identify the target population (Supplementary Fig. 15). Data were acquired and analysed with the CytExpert 2.6 software (Beckman Coulter). The specific growth rate (k; day−1) was estimated via linear regression analysis of ln(Cellt/Cell0) versus time (day) over an apparent exponential growth phase. Here, Cellt is the cell concentration (in cells per ml) at sampling time t (day).
13C-bicarbonate labelling experiments and isotope analysis
To probe for autotrophic carbon fixation during MISO growth conditions, 13C-labelled bicarbonate (98 atom% 13C; Sigma Aldrich) was added to ferrihydrite-incubated cultures receiving dissolved sulfide (1 mM) or FeS (ca. 1 mM S equivalent), to reach a 10 atom% 13C in the inorganic carbon pool. The dissolved sulfide or solid phase FeS were spiked in the same frequency as for the growth experiment. Abiotic controls for each culture were set up using autoclaved inoculum. To detect 13C content in bulk biomass and in single cells, subcultures were sampled, fixed by formaldehyde (2.3% final concentration), and analysed using elemental analyser-isotope ratio mass spectrometry (EA-IRMS) and NanoSIMS. For EA-IRMS, 1.5 ml of fixed samples that included ferrihydrite and cells were pelleted by centrifugation and washed with MilliQ water, followed by overnight treatment by 0.1 M HCl to remove residual carbonates. The dried cells attached to ferrihydrite particles were weighed (4–6 mg) and transferred to tin cups. Bulk cell carbon isotope ratios (13C:12C) were measured by EA-IRMS (Delta V Advantage) coupled by a ConFlo IV interface to an elemental analyser (EA-Isolink, all Thermo Finnigan). Sample 13C contents were calculated as atomic percentage of 13C in total carbon, following 13C atom% = 13C/(13C + 12C) × 100%. The analytical precision of replicate analyses of isotopically homogeneous international standards was ±0.0001% for 13C atom% measurements.
For NanoSIMS analysis, 0.1 ml formaldehyde-fixed samples that included ferrihydrite and cells were mixed with dithionite solution as described above and incubated for 2 h. After complete dissolution of ferrihydrite, 400 µl of the suspension was transferred onto gold-coated polycarbonate filters (GTTP type, 0.2 µm pore size, Millipore). The filters were gold-coated by physical vapour deposition, utilizing an Agar B7340 sputter coater (Agar Scientific) equipped with an Agar B7348 film thickness monitor (Agar Scientific) for precise adjustment of the coating thickness (150 nm). The filters were incubated for 2 h in 0.1 M HCl to remove residual carbonates and then washed twice in MilliQ water and then air-dried. Filter sections were attached to antimony-doped silicon wafers (7.1 ×7.1 mm, Active Business Company) with a commercially available glue (SuperGlue Loctide).
NanoSIMS measurements were carried out on a NanoSIMS 50 L instrument (Cameca) at the Large-Instrument Facility for Environmental and Isotope Mass Spectrometry at the University of Vienna. Prior to data acquisition, analysis areas were pre-conditioned in situ by rastering a high-intensity, slightly defocused Cs+ ion beam for removal of surface adsorbates and establishment of the steady state secondary ion signal intensity regime with minimum sample erosion. For this purpose, the following sequence of high and extreme low Cs+ ion impact energy (EXLIE) was applied: high energy (16 keV) at 100 pA beam current to a fluence of 5 × 1014 ions cm−2; EXLIE (50 eV) at 400 pA beam current to a fluence of 5 × 1016 ions cm−2; high energy to an additional fluence of 2.5 × 1014 ions cm−2. Data were acquired as multilayer image stacks by scanning of a finely focused Cs+ primary ion beam with 2 pA beam current at approximately 80 nm physical resolution (probe size) over areas between 60 × 60 and 62 × 62 µm2 with 512 × 512 pixel and 1,024 × 1,024 pixel image resolution and a per-pixel dwell time of 5 ms and 1.5 ms, respectively. The detectors of the multicollection assembly were positioned for parallel detection of 12C2−, 12C13C−, 12C14N−, 31P− and 32S-secondary ions. Secondary electrons were detected simultaneously for gaining information about the sample morphology and topography. The mass spectrometer was tuned to achieve a mass resolving power ((MRP) = M/ΔM) of >10,000 for detection of C2− secondary ions.
Measurement data were processed using the WinImage software package provided by Cameca (WinImage V4.8) and the OpenMIMS plugin in the image processing package ImageJ (V1.54p). Prior to data evaluation, images were corrected for detector dead-time and positional variations emerging from primary ion beam and/or sample stage drift. Carbon isotope composition images displaying the 13C/(12C + 13C) isotope fraction, given in atom percent (atom%), were inferred from the C2− secondary ion signal intensity distribution images via per-pixel calculation of 13C12C−/(2 × 12C2− + 12C13C−) intensity ratios. For numerical data evaluation, regions of interest, referring to individual cells, were manually defined on the basis of the 12C14N− and 31P− secondary ion maps as indicators of biomass and verified by the topographical/morphological appearance in the secondary electron images. Biomass aggregates, in which an unambiguous identification of single cells was not feasible, were rejected.
Cells were assessed as being significantly enriched in 13C after incubation in the presence of 13C-bicarbonate if (1) the 13C isotope fraction value was higher than the mean plus 3 standard deviations (σ) of the values determined on the cells from the control (on day 0) and (2) the statistical counting error (3σ, Poisson) was smaller than the difference between the considered 13C enriched cell and the mean value measured on the cells from the control. The Poisson error was calculated from the secondary ion signal intensities (given in counts per region of interest) via
$${\sigma }_{{\rm{Pois}}}=1/{\left(2\times \genfrac{}{}{0ex}{}{12}{}{{{\rm{C}}}_{2}}^{-}+\genfrac{}{}{0ex}{}{12}{}{\rm{C}}\genfrac{}{}{0ex}{}{13}{}{{\rm{C}}}^{-}\right)}^{2}\times \sqrt{\left({\left(\genfrac{}{}{0ex}{}{12}{}{{{\rm{C}}}_{2}}^{-}\right)}^{2}\times \genfrac{}{}{0ex}{}{12}{}{\rm{C}}\genfrac{}{}{0ex}{}{13}{}{{\rm{C}}}^{-}+{\left(\genfrac{}{}{0ex}{}{12}{}{\rm{C}}\genfrac{}{}{0ex}{}{13}{}{{\rm{C}}}^{-}\right)}^{2}\times \genfrac{}{}{0ex}{}{12}{}{{{\rm{C}}}_{2}}^{-}\right)}$$
On the basis of these two criteria, all individual cells measured in the 13C incubated sample showed a significant enrichment in 13C.
RNA-seq and transcriptomics
D. alkaliphilus cultures grown under five incubation conditions (as described in ‘Cultivation of D. alkaliphilus DSM 19089’), each in four replicates, were used for comparative transcriptomic analysis. Cultures (30 ml) showing metabolic activity (for example, Fe(ii) production, sulfide consumption or production) were collected in the middle to late stage of incubation experiments. Cells were collected by centrifuging (12,857g; room temperature) under anoxic condition using oak ridge tubes (Thermo Fisher Nalgene) with replacement O-rings for sealing cap (Thermo Fisher Nalgene). The cell pellets were resuspended with 1.5 ml supernatant, and distributed to three lysis matrix E tubes (MP Biomedicals), each with approximately 0.5 ml. The collected cells were immediately frozen with liquid N2, and stored at −80 °C before subsequent analysis. The total nucleic acids were extracted following a phenol-chloroform protocol as described previously99,100. In brief, the sample was lysed for 30 s at a speed of 5.5 m s−1, after mixing with hexadecyltrimethylammonium bromide extraction buffer and phenol-chloroform-isoamyl alcohol (25:24:1) (pH 8.0). The aqueous phase was extracted by centrifugation, and the phenol within was removed by mixing with chloroform-isoamyl alcohol (24:1). The total nucleic acids in the aqueous phase were then precipitated with polyethylene glycol 6000, followed by centrifugation. The pelleted nucleic acids were washed with ice-cold ethanol and dried before resuspension in diethyl pyrocarbonate-treated water. DNA from the total nucleic acids were removed using the TURBO DNA-free kit (Thermo Fisher Scientific).
RNA-sequencing was performed at the Joint Microbiome Facility of the Medical University of Vienna and the University of Vienna (JMF) under project IDs JMF-2311-14 and JMF-2405-05. Sequencing libraries were prepared from rRNA depleted (Ribo-Zero Plus rRNA Depletion Kit, Illumina) RNA samples (NEBNext Ultra II Directional RNA Library Prep Kit for Illumina, New England Biolabs) and sequenced in 2× 100 bp paired-end mode (NextSeq 6000, Illumina), yielding 74.2–303.7 million raw reads per sample. Individual read libraries were quality checked using fastQC v0.12.1 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and quality statistics were merged using multiQC v1.21 (ref. 101). Adapters were trimmed and phiX contamination was removed using BBDuk (part of BBMap v39.06). Reads were k-trimmed from the right with a kmer of 21, minimum kmer of 11 and hamming distance of two along with the tpe and tbo options. Quality trimming was performed from the right with a q-score of 28 to a minimum of 50 bases in length (https://sourceforge.net/projects/bbmap/). The quality filtered reads were aligned to the reference genome of D. alkaliphilus (NC_014216.1) using BBMap with a mapping identity of 99% and with ambiguous reads assigned to the best location (that is, counted only once for duplicated genes). FeatureCounts (part of SubRead 2.0.6 (ref. 102)) with reverse-stranded and –countReadPairs were used to generate counts tables with the resulting alignments based on gene call locations by prodigal v2.6.3 (ref. 103). Counts tables were analysed using DESeq2 release 3.19 (ref. 104) to calculate the RPKM and to determine statistical significance of differential transcription between treatment groups. All P values are adjusted for multiple comparisons using the Benjamini–Hochberg method105.
Quantitative PCR with reverse transcription (RT–qPCR) was performed to verify the upregulated transcription for the MHC gene DA_402 under iron-reducing conditions. Primers DA_402_998F (5′-TTCCCAATCGGGGCGAATAC-3′) and DA_402_1081R (5′-TGGCCTCGGTATAGAGGGTC-3′) were used to target DA_402. Primers recA_79F (5′-TTCGGCAAAGGCTCCATCAT-3′) and recA_221R (5′-TCCGGCCCATATACCTCGAT-3′) were used to quantify the transcription level of the house-keeping gene recA (DA_1926) encoding the DNA recombination protein. Primers for both genes were newly designed using Primer-Blast106. For RT–qPCR, DNA-free RNA was first reverse transcribed to cDNA using SuperScript III reverse transcriptase according to the manufacturer’s instructions. The absolute abundance of transcripts from DA_402 and recA were quantified by quantitative PCR using cDNA as a template. Purified PCR products of gene DA_402 and recA amplified from genomic DNA of D. alkaliphilus were used as quantitative PCR standards. The PCR reactions were prepared in triplicates and run at 95 °C for 3 min, followed by 40 cycles of 95 °C for 30 s, 60 °C for 30 s, and 72 °C for 45 s, on the Thermal Cycler with CFX96 Real-Time System (Bio-Rad). The RT–qPCR data were acquired and analysed using CFX Maestro software (Bio-Rad). The transcription level of DA_402 was compared between treatments after normalization with that of recA. The statistical significance of differential transcription between treatments were determined via Student’s t-test.
Structure prediction and phylogenetic analysis of multi-haem c-type cytochromes in D. alkaliphilus
D. alkaliphilus proteins with more than one haem-binding motifs (CXnCH; n = 2 to 5) were considered MHCs107. The haem-binding motifs in protein sequences were counted using regex expressions in the Python re package. The subcellular localization of all putative MHCs (n = 46) from D. alkaliphilus was predicted using PSORTb v3.0 (ref. 108) and DeepLocPro v1.0 (ref. 81). Prediction from DeepLocPro was used for the proteins for which PSORTb returned ‘Unknown’. The transcription levels of MHCs were compared between different incubation experiments on the basis of RPKM values. The statistical significance of differential transcription was assessed as described in the ‘RNA-Seq and transcriptomics’ chapter. The most highly transcribed extracellular MHC (DA_402) during MISO was further selected for structure prediction and phylogenetic analysis. The structure of the DA_402 monomer and oligomer were predicted using AlphaFold2 v2.3.2 at the COSMIC2 science gateway. The leading signal peptide, predicted using SignalP 5.0 (ref. 109), was cleaved from the protein sequence before structure modelling. For comparison, the cryo-EM structure of OmcS from G. sulfurreducens was retrieved from the Protein Data Bank (PDB) database (6EF8). The protein sequence of DA_402 was aligned to OmcS using the T_coffee alignment tool110. The structure-structure similarity between DA_402 and OmcS was calculated using an online TM-align tool and DaliLite.v5 (ref. 111,112,113). The haem-binding sites in DA_402 and OmcS were visualized using MacPyMOL v.1.7.4 (https://pymol.org). The haem was docked to the target haem-binding site in DA_402 using AutoDockTools v1.5.7 (ref. 114) and AutoDock Vina 1.1.2 (ref. 115). To conduct phylogenetic analysis of DA_402, homologues of DA_402 were retrieved from the KEGG database using Blastp with an E value of 0.01. The retrieved homologues were then de-replicated with CD-HIT at 70% identity, aligned with Muscle, trimmed with trimAl (–gt 0.1). The resulting sequence alignment was used to reconstruct the maximum-likelihood tree using RAxML v8.2.12. The clustering pattern and decoration of the tree were performed using iTOL v6 (ref. 116).
Environmental distribution of Desulfurivibrionaceae with genomic potential of MISO
The metabolic potential of members belonging to the Desulfurivibrionaceae family was analysed using publicly available genomes recovered from different environments. Metagenome-assembled genomes (MAGs) classified as Desulfurivibrionaceae were obtained from GTDB r214 (n = 121), NCBI (n = 9), JGI IMG (n = 68) and GMGC (n = 7). The environmental origins of these genomes were retrieved from the metadata in the respective databases (Supplementary Table 8). The taxonomy of collected genomes was assigned using GTDB-tk version 2.3.2 with database release 214 (ref. 117). The phylogenomic tree of Desulfurivibrionaceae was reconstructed from a concatenated alignment of 120 single-copy genes with FastTree v2.1.10 (ref. 66). The protein-coding genes in the genomes were called using Prodigal v2.6.3 (ref. 103), and the resulting proteomes were screened for proteins involved in dissimilatory sulfide oxidation (DsrAB) and iron oxides reduction (that is, OmcS, OmcZ, porin–cytochrome complex and OmcE). DsrAB was detected using HMMs and the phylogenetic framework established in this study, while proteins involved in dissimilatory iron reduction were identified with HMMs from FeGenie86. Additional proteins likely involved in dissimilatory reduction of iron oxides—that is, extracellular MHC DA_402 and PilA—were retrieved from Desulfurivibrionaceae genomes by hmmsearch or BLASTP. Homologues of PilA were extracted by searching TIGR02532 HMM model against the Desulfurivibrionaceae genomes using hmmsearch with –cut_ga option. For DA_402, homologues were collected from the Desulfurivibrionaceae genomes using BLASTP with an E value of 1e-10, followed by prediction of the subcellular localization and counting of haem-binding sites. The extracellular homologues containing multi-haem-binding sites (n > 3) were then placed into a reference tree created through phylogenetic analyses of DA_402 (see above) with the RAxML evolutionary placement algorithm (EPA). The alignment for EPA was generated using MAFFT v7.407 with –add option. The homologues that were placed with accumulated probability over 0.95 to the OmcS-like clade were considered as functional orthologs of DA_402. For visualization purposes, Desulfurivibrionaceae genomes (n = 119) encoding both dissimilatory iron and sulfur metabolism were de-replicated on the basis of relative evolutionary divergence (RED). RED was calculated for each internal node of the Desulfurivibrionaceae phylogenomic tree following the procedure described previously118. The tree was then collapsed at a RED value of 0.9 and one representative was chosen randomly from the collapsed clades, yielding 53 representative members that were visualized in the tree.
Statistics and reproducibility
The physiological experiments showing the ability of ferrihydrite-incubated D. alkaliphilus to oxidize formate (Fig. 3a), FeS (Fig. 3b), 1 mM sulfide (Fig. 3c,d,g) or ~50 µM sulfide (Fig. 3h) were repeated independently at least three times, all yielding consistent results. The sulfide removal kinetic experiments at low sulfide concentration were replicated independently for two times, and all results are present in the Extended Data Fig. 1. The experiment showing the transformation of sulfide and ferrihydrite with periodic supply of ~50 µM sulfide was performed independently twice, and both yielded similar results. The growth experiments of ferrihydrite-incubated cells with periodic addition of 1 mM sulfide (Fig. 4a), FeS (Fig. 4b) or 50 µM sulfide (Fig. 4c), and the experiment with nitrate and sulfide (Fig. 4d) were conducted once with three biological replicates per treatment and control. The 13C-bicarbonate labelling experiment and bulk 13C analysis of cells incubated with ferrihydrite and either dissolved sulfide (Fig. 4e) or FeS (Fig. 4f) were independently repeated for two times, both yielding comparable outcomes. Samples for NanoSISM analysis were chosen randomly among biological replicates collected at day 0 and 5, and representative field views are present in Fig. 4g–i. Transcriptomic analysis of cells growing under five different conditions was conducted once, with four biological replicates per condition (Fig. 5 and Extended Data Fig. 4).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.