
Photophysical properties of the guests in crystal
Three compounds r-MOP, s-MOP, and MOP were designed as the guests, then purified and characterized by the 1H NMR and single-crystal X-ray diffraction (Supplementary Figs. 1–3 and Supplementary Table 1). Photo-activated luminescent properties of the r-MOP, s-MOP, and MOP in crystal were measured. r-MOP, s-MOP, MOP in crystals showed blue emission under a 365-nm lamp on, and no phosphorescence can be observed after the remove of a 365 nm lamp (Supplementary Fig. 6). Notably, blue emission from three crystals turned orange after the irradiation of 365-nm lamp, followed by green UOP for several seconds (Supplementary Fig. 6). The photoluminescence spectra of three crystals have strong emission peak at 409 nm and weak shoulder peak at 496 nm at the beginning, then the two peaks decreased rapidly and a new peak at 599 nm appeared with prolonging the excitation time (Supplementary Figs. 7a–c and Supplementary Table 2). EPR spectra of r-MOP, s-MOP, and MOP crystals before and after the irradiation of a 365 nm lamp revealed that the free radicals were produced after the irradiation of 365 nm (Supplementary Fig. 8). The emission peak at 599 nm belongs to the emission of free radicals. In addition, phosphorescence spectra of three crystals exhibited strong peak at 501 nm and a shoulder peak at 536 nm, phosphorescence intensity decreased with the irradiation of 365 nm UV light (Supplementary Fig. 7d–f). Interestingly, phosphorescence emission color turned from green to yellow with further photo-activation (Supplementary Figs. 7–10 and Supplementary Table 3).
To investigate the mechanism of photo-activated UOP in crystals, we analyzed the single crystals of r-MOP, s-MOP, and MOP. Each molecule is fixed by multiple hydrogen bonding interactions (C-H⋯O, C-H⋯P, C-H⋯π, C-H⋯H-C) (Supplementary Figs. 11 and 12). After the photo-activation, the number of hydrogen bonding C-H⋯O and C-H⋯π interactions are increasing (Supplementary Figs. 11 and 12). The photo-activated UOP can be ascribed to the increasing number of hydrogen bonding interaction to restrict the molecular motion. The absorption spectra of r-MOP, s-MOP, MOP in crystal exhibited unchangeable at 308 nm, and significant enhancement ranging from 375 nm to 600 nm after the photo-activation (Supplementary Fig. 13). The photo-activated phosphorescence spectra showed a large overlap with their photo-activated absorption, suggesting that the attenuated phosphorescence intensity and lifetime can be attributed to the energy transfer from the neutral molecule to radical pairs.
Photophysical properties for r-MOP or r-MOP-1 doped into PMMA film
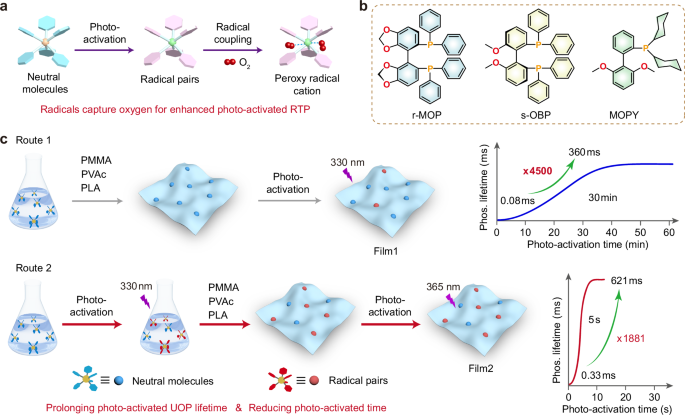
We discovered that r-MOP, s-MOP, MOP in the dilute chloroform solution (1 × 10−5 M) also exhibited the photo-activated photoluminescence emission (Supplementary Fig. 15). Therefore, we speculate that r-MOP, s-MOP, and MOP in the single-molecule state can also achieve photo-activated UOP. To validate our hypothesis, taking r-MOP for an example, 1 wt.% r-MOP in PMMA film was prepared. As expected, it demonstrated photo-activated photoluminescence and phosphorescence upon the 330 nm UV irradiation (Fig. 2a and Supplementary Fig. 15). In addition, phosphorescence intensity of 1 wt.% r-MOP in PMMA was slowly enhanced with the photo-activation time, it needs about 42 min to reach saturation (Fig. 2a, b and Supplementary Fig. 16). As shown in Fig. 2c, no emission of 1 wt.% r-MOP in PMMA film can be observed by the naked eye before the photo-activation. Under a 365-nm lamp on, the emission color of PMMA films turned from orange to yellow, then bluish-green, with the emission peaks from 560 nm to 490 nm after photo-activated by 302 nm (Fig. 2c and Supplementary Figs. 17 and 18). And photo-activated PMMA films emitted bright cyan afterglow under 365 nm lamp off, the cyan afterglow was prolonged with photo-activated time from 0 min to 30 min (Fig. 2c and Supplementary Fig. 17). Phosphorescence lifetime of 1 wt.% r-MOP in PMMA film increased from 0.08 ms to 360 ms (Fig. 2d). Both phosphorescence intensity and lifetime remained almost unchangeable after the further photo-activation (Fig. 2b, d). With excitation light intensity increased from 0.19 to 3.16 mW/cm2, the photo-activation time of 1 wt.% r-MOP in PMMA was shortened gradually (Supplementary Fig. 20). PMMA films doped with r-MOP at different doped concentrations demonstrated similar phosphorescence spectra (Supplementary Fig. 19). The photo-activated phosphorescence lifetime and efficiency initially increased, followed by a subsequent decrease with the doped concentrations (Supplementary Figs. 21 and 22 and Supplementary Table 4). Phosphorescence intensity and lifetime can recover after kept in dark for 50 min.
Fig. 2: Photophysical properties of r-MOP or r-MOP-1 doped into PMMA film.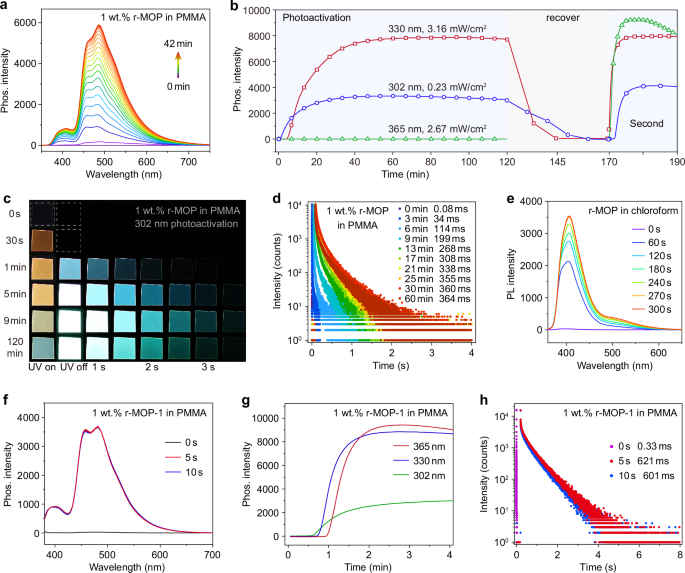
a Phosphorescence (Phos.) spectra (Interval: 2 min, 3.16 mW/cm2) at different lengths of irradiation time excited by 330 nm lamp under ambient conditions. b The phosphorescence intensity of 1 wt.% r-MOP in PMMA film monitoring 484 nm under the irradiation of 302, 330, or 365 nm, the recovery of phosphorescence, and the photo-activation for second time. c Photographs of 1 wt.% r-MOP in PMMA film taken under 365 nm UV lamp (39.9 mW/cm2) on and off after photo-activated by 302-nm UV lamp (4.04 mW/cm2) with different irradiation time. d Lifetime decay profiles of 1 wt.% r-MOP in PMMA film excited by 365 nm monitoring 484 nm after photo-activated by 302 nm. e Steady-state photoluminescence (PL) spectra of r-MOP in chloroform photo-activated by 302 nm. f Phosphorescence spectra of 1 wt.% r-MOP-1 in PMMA films photo-activated by 365 nm. g The curves of phosphorescence (Phos.) intensity with the irradiation time monitoring 484 nm photo-activated by 302, 330, or 365 nm. h Lifetime decay profiles of 1 wt.% r-MOP-1 in PMMA film excited by 365 nm monitoring 484 nm after photo-activated by 365 nm.
Notably, 1 wt.% r-MOP in PMMA films show no phosphorescence emission after the initial photo-activation by 365 nm (Fig. 2b). Impressively, the photo-activated PMMA films can be rapidly photo-activated by 302, 330, or 365 nm UV light for the second time (Fig. 2b). Only for 4 min is required that phosphorescence spectra of 1 wt.% r-MOP in PMMA film reach the saturation after photo-activated by 330 nm lamp for the second time (Supplementary Fig. 23a). Besides 330 nm lamp, 1 wt.% r-MOP in PMMA film can be quickly photo-activated by 365 nm for the second time (Supplementary Fig. 23b). Notably, the phosphorescence lifetime of 1 wt.% r-MOP in PMMA film was obviously prolonged from 360 ms to 529 ms after photo-activated by 302 nm lamp for the second time, and exhibited a longer phosphorescence lifetime of 615 ms after photo-activated by 365 nm for the second time (Supplementary Fig. 23c, d and Supplementary Table 5). The cyan afterglow can last for over 5 s (Supplementary Fig. 24). In a word, 1 wt.% r-MOP in PMMA film demonstrated similar phosphorescence spectra, but short photo-activation time and longer phosphorescence lifetime after photo-activated by 302, 330, or 365 nm for the second time. compared with that of for the first photo-activation. It can be ascribed to the production of radical during the first photo-activation, which accelerates the photo-activation process for the second time and enhances the phosphorescence emission simultaneously.
To further verify the enhancement of radical on photo-activated UOP, a series of control experiments were conducted. Firstly, r-MOP in chloroform solution was photo-activated for 4.5 min, then its photoluminescence intensity increased with the photo-activated time to reach saturation (Fig. 2e), finally doped into PMMA to prepare 1 wt.% r-MOP-1 in PMMA film. Impressively, 1 wt.% r-MOP-1 in PMMA films exhibited quickly photo-activated phosphorescence emission under the irradiation of 365 nm (Fig. 2f, g and Supplementary Fig. 25a). 1 wt.% r-MOP-1 in PMMA films showed blue emission under 365 nm lamp on, and cyan afterglow under 365 nm lamp off, which can last for 7 s after photo-activated by 365 nm for just only 5 s (Supplementary Fig. 25b), with long phosphorescence lifetime of 621 ms (Fig. 2h). The phosphorescence lifetimes decreased after long irradiation of 365 nm (Fig. 2f). In addition, photo-activated UOP of 1 wt.% r-MOP-1 in PMMA recovered after kept in dark for 15 min (Supplementary Fig. 25c). To investigate the influence of pre-solution irradiation duration on the phosphorescence lifetime and photo-activated time, r-MOP in chloroform was photo-activated by 302 nm for 1, 2, 3, 4, 5, and 6 min, respectively, then doped into PMMA solution to prepared 1 wt.% r-MOP in PMMA-X min (X = 1, 2, 3, 4, 5, 6) films. All films can be photo-activated by 365 nm rapidly with short photo-activation time of 3 s or 5 s (Supplementary Fig. 26). But photo-activated phosphorescence lifetimes were prolonged from 504 ms to 668 ms with pre-solution irradiation duration from 1 min to 5 min, then decreased with pre-solution irradiation time at 6 min (Supplementary Fig. 27 and and Supplementary Table 6). As shown in Supplementary Fig. 28, the cyan afterglow of 1 wt.% r-MOP in PMMA-5 min films can last for over 6 s, exhibiting the longest phosphorescence emission.
Universality of the approach for photo-activated UOP
To investigate the generality of our approach for photo-activated UOP, a series of parallel experiments were carried out. To clarify the influence of guest on photo-activated UOP, s-MOP, MOP, MOPY, and s-OBP were selected as the guests. The photo-activated phosphorescence spectra and lifetime of PMMA films doped with 1 wt.% s-MOP or 1 wt.% MOP are similar with that of 1 wt.% r-MOP, suggesting that the chirality of the guest in PMMA have little influence on photo-activated UOP (Fig. 3a and Supplementary Figs. 29 and 30). PMMA films doped with 1 wt.% MOPY, or 1 wt.% s-OBP also demonstrated photo-activated UOP (Fig. 3a). Interestingly, their photo-activated phosphorescence spectra exhibited an obvious redshift from 483 nm to 554 nm, with UOP emission from cyan to green, then yellow (Fig. 3a and Supplementary Figs. 30 and 31). In addition, their phosphorescence lifetimes were dramatically enhanced after the photo-activation (Fig. 3b and Supplementary Fig. 32), and recovered in the dark (Supplementary Figs. 33 and Supplementary Tables 7 and 8).
Fig. 3: Photophysical properties of PMMA doped with different guests, and different guests doped into PVAc film, PLA film, or PLA glass under ambient conditions.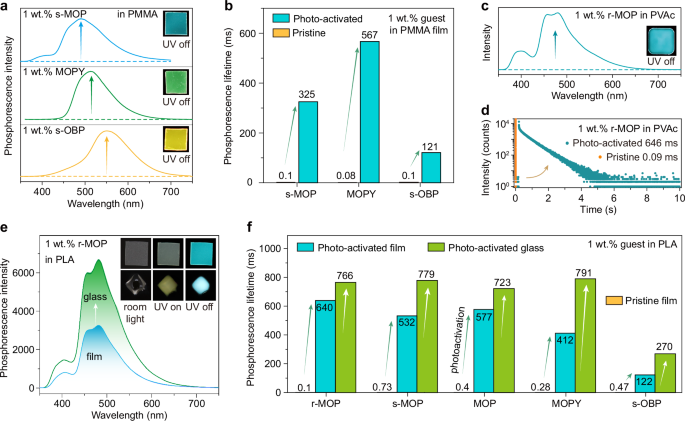
a Phosphorescence spectra of 1 wt.% s-MOP in PMMA, 1 wt.% MOPY in PMMA, and 1 wt.% s-OBP in PMMA before (dash lines) and after photo-activated (solid lines) by 330, 330, and 270 nm, respectively. Insets: Photographs of PMMA films photoactivated by corresponding excitation source taken under 365 nm lamp off. b Phosphorescence lifetimes of 1 wt.% s-MOP in PMMA, 1 wt.% MOPY in PMMA, and 1 wt.% s-OBP in PMMA before and after the photo-activation. c Phosphorescence spectra and d phosphorescence lifetimes of 1% r-MOP in PVAc before and after photo-activated by 330 nm. Insets: corresponding photograph of photo-activated PVAc films taken under 365 nm lamp off. e Phosphorescence spectra of 1% r-MOP in PLA film (cyan line) and glass (green line) after photo-activated by 330 nm. Insets: the corresponding photographs taken under the room light, 365 nm lamp on and off. f Phosphorescence lifetimes of PLA films doped with r-MOP, s-MOP, MOP, MOPY, and s-OBP at the concentration of 1 wt.% before (orange columns) and after the photo-activation (cyan columns), and corresponding melting PLA glass after the photo-activation (green columns).
To explore the impact of the host on photo-activated UOP, poly(vinyl acetate) (PVAc) and polylactic acid (PLA) were chose as the hosts. As anticipated, PVAc or PLA doped with different guests also showed photo-activated RTP. Both photo-activated photoluminescence and phosphorescence spectra of PVAc and PLA films doped with different guests are similar to that of PMMA doped with the corresponding guest (Fig. 3c and Supplementary Figs. 42–45), except for strong shoulder peak at 456 nm in photo-activated photoluminescence spectra of PVAc and PLA films doped with r-MOP, s-MOP, MOP. The photo-activation time of PVAc and PLA films were markedly reduced, and phosphorescence lifetimes were considerably prolonged compared with that of PMMA films doped with the corresponding guest (Fig. 3c–f and Supplementary Figs. 46–50, Supplementary Tables 11 and 12). PLA films showed a certain crystalline degree, while PLA glass exhibited amorphous state (Supplementary Fig. 51). Surprisingly, phosphorescence intensity and lifetimes were further enhanced after PLA films were melted into PLA glass (Fig. 3e, f). In PLA glass, the polymer chains are packed more densely, providing a more rigid environment that suppresses non-radiative transitions of the luminescent molecules. The preparation of PLA glass requires high-temperature conditions, which more thoroughly removes oxygen, moisture, and solvents from the PLA polymer matrix. The photoluminescence and phosphorescence spectra of PLA glass are similar to that of PLA films, but need longer photo-activation time (Supplementary Figs. 52–57 and Supplementary Table 13).
Mechanism investigation for the photo-activated UOP
To get an insight into the mechanism of photo-activated UOP, EPR spectra of 1 wt.% r-MOP in PMMA films and r-MOP in crystal were measured, the result reveals that free radicals are produced after the photo-activation of r-MOP molecule (Fig. 4a). The absorption of r-MOP in crystal showed a peak at 308 nm, which may be the absorption of the neutral r-MOP molecules (Fig. 4b). The enhanced absorption ranging from 375 nm to 600 nm after the photo-activation might be attributed to the radicals. The photo-activated phosphorescence spectrum of r-MOP in crystal showed a large overlap with its absorption (Fig. 4b), and phosphorescence intensity decreased with the long photo-activation (Supplementary Fig. 8), suggesting the energy transfer between the neutral molecules to the radical pairs. By contrast, thick 1 wt.% r-MOP in PMMA film and 1 wt.% r-MOP-1 in PMMA film with the thickness of 0.131 mm exhibit stable and strong absorption at 302 nm, and incremental absorption at 355 nm with the 302-nm UV irradiation (Fig. 4c and Supplementary Fig. 66a). 1 wt.% r-MOP-1 in PMMA film showed more intense absorption at 355 nm, owing to high concentration of radical in PMMA film. The absorption of 1 wt.% r-MOP in PMMA film with a quarter thickness (0.033 mm) is weak at 302 nm, decreases after photo-activated by 302 nm (Supplementary Fig. 66b), which attributes to the transfer of partial neutral molecules to radical pairs. The photo-activated phosphorescence spectra of 1 wt.% r-MOP in PMMA films demonstrated a large Stokes shift (Fig. 4c), therefore, the energy transfer from the neutral molecules to the radical pairs can be effectively impeded. Therefore, 1 wt.% r-MOP in PMMA films present photo-activated UOP with high photo-stability (Fig. 2b). EPR spectra of PMMA films doped with different guests and r-MOP doped into PVAc or PLA indicated the generation of the radical pairs after the photo-activation (Supplementary Figs. 67 and 68). Their photo-activated phosphorescence spectra showed negligible overlap with their absorption spectra (Supplementary Figs. 69 and 70), suggesting that the energy transfer was hindered for PMMA, PVAc or PLA films.
Fig. 4: Mechanism investigation of radical-enhanced photo-activated UOP from doped polymer films.
a Electron paramagnetic resonance (EPR) spectra of r-MOP in crystal and 1 wt.% r-MOP in PMMA before and after the photo-activation. The absorption of (b) r-MOP in crystal and c 1 wt.% r-MOP in PMMA films photo-activated by 365 nm and 302 nm with different irradiation time, respectively. d Phosphorescence spectra of r-MOP in dilute chloroform solution (1 × 10−5 M) and 1 wt.% r-MOP in PMMA film at 77 K before and after photo-activated by 330 nm at room temperature. e Natural transition orbitals (NTOs) contributing to the lowest-energy triplet transitions of r-MOP monomer and s-OBP monomer. f Lifetime decay profiles of photo-activated 1 wt.% r-MOP in PMMA films coated with oxygen barrier excited by 365 nm monitoring 484 nm. g Proposed mechanism of quickly photo-activated UOP. Note: S0 the ground state, S1 the lowest singlet excited state, Sn singlet excited states, IC internal conversion, ISC intersystem crossing, Abs. absorbance, Fluo. fluorescence, Phos. phosphorescence, D0 ground doublet state, D1 and Dn excited doublet states, ET energy transfer.
To further probe the mechanism of photo-activated UOP, we measured phosphorescence spectra of 1 wt.% r-MOP in PMMA films and r-MOP in dilute chloroform solution (1 × 10−5 M) at 77 K before and after photo-activated at room temperature (Fig. 4d). The phosphorescence spectra of 1 wt.% r-MOP in PMMA films is similar with that of r-MOP in dilute chloroform solution (1 × 10−5 M) at 77 K, suggesting the phosphorescence originates from the isolated neutral molecule. Moreover, we calculated the natural transition orbitals (NTOs) of contributing to the lowest-energy triplet transitions of r-MOP monomer and s-OBP monomer (Fig. 4e). NTOs of r-MOP monomer are located on a phenyl group of biphenyl group, adjoining phosphorus and oxygen atoms. NTOs of s-OBP monomer bestrewed the whole biphenyl group. To concretize the energy level of frontier molecular orbital, the energy levels of the HOMO of r-MOP and s-OBP were determined by the ultraviolet photoelectron spectroscopy (UPS) method and cyclic voltammetry (CV) study (Supplementary Figs. 71 and 72). The energy level of the HOMO of s-OBP is lower than that of r-MOP. Hence, the phosphorescence emission color was easily tailored by modifying the skeleton of biphenyl group, owing to the enhanced conjugation.
To validate the photo-activated UOP from the consumption of oxygen in polymer upon continuous ultraviolet illumination, we adopted indirect method to verify the generation of singlet oxygen (1O2) under UV-light irradiation. The absorption peaks of ADMA of gradually decreased and eventually disappeared upon the UV irradiation to confirm the production of singlet oxygen (1O2) (Supplementary Fig. 74). The energy transfer (ET) between the triplet state of r-MOP and the ground state of molecular oxygen (3O2) can lead to the generation of an electronically excited state of molecular oxygen, i.e. singlet oxygen (1O2) upon photo-excitation. In addition, the triarylphosphine radical cation Ar3P•+ can capture the oxygen by through radical coupling with O2 to afford the peroxy radical cation Ar3P+–O–O•31,32,33. The synergy of UV irradiation and the radical coupling of triarylphosphine radical cation consumed the oxygen to accelerate the photo-activation and prolong the photo-activated phosphorescence lifetimes. 1 wt.% r-MOP in PMMA can also produce photo-activated UOP in vacuum or nitrogen, exhibiting longer photo-activated lifetime, and recovered slowly in nitrogen (Supplementary Figs. 75 and 76 and Supplementary Table 17). In addition, the photo-activated UOP of 1 wt.% r-MOP in PMMA films and 1 wt.% r-MOP-1 in PMMA films did not recover after coated with oxygen barrier (poly(vinyl alcohol-co-ethylene)) (Fig. 4f and Supplementary Fig. 77). Therefore, the photo-activated UOP of PMMA, PVAc, or PLA films, and PLA glass originated from the consumption of oxygen in polymers (Fig. 4g).
Applications of photo-activated UOP films
Given its excellent photo-activated UOP performance of PMMA films doped with different guests, we explored its potential applications in the fields of information storage and anti-counterfeiting. As shown in Figs. 5a, 1 wt.% r-MOP in PMMA film showed no emission at the beginning, a pattern quick response code was easily printed on the 1 wt.% r-MOP in PMMA film via the photo-printing technology of 302 nm UV light (Supplementary Fig. 78a). Blue and cyan quick response codes were presented under a 365 nm lamp on and off, respectively. After the pattern was placed in the air over 1 year, its emission did not change, exhibiting great potential in information storage. Then the pattern hid by the irradiation of 302 nm lamp without mask (Supplementary Fig. 78a). The film is kept in dark for 50 min, the photo-activated UOP can recover and the photoluminescence image remained unchangeable (Fig. 5a). Other photo-activated pattern can be prepared by using specified mask upon the film (Supplementary Fig. 78a), and erased by leaving in dark for 50 min. Based on writing, reading and erasing procedure of 1 wt.% r-MOP in PMMA film, various afterglow patterns were written by photo-activation successively and erased by leaving in dark for 50 min onto the same substrate (Fig. 5b and Supplementary Fig. 78b). In addition, multicolor phosphorescence displays were achieved by the photo-activation of PMMA films doped different guests (Fig. 5c, Supplementary Figs. 79 and 80a). In view of irradiation time-dependent photo-activated UOP, the letters “RTP” and the numbers “2024” were printed on the same film with the photo-activation time of 18 min and 4 min, respectively (Supplementary Fig. 80b). Interestingly, they exhibited blue “RTP” and red “2024” under a 365-nm lamp on, then presented cyan “RTP” and “2024”, followed by green “RTP” under a 365-nm lamp off (Fig. 5d). Besides PMMA films, photo-activated UOP PVAc film and PLA glass can also be applied for anti-counterfeiting and decoration. Transparent 1 wt.% MOPY in PVAc film on the bottle was not easily perceived, but cryptographic information appeared after the photo-activation, exhibiting the practical application in anti-counterfeiting (Fig. 5e and Supplementary Fig. 80c). The shells were covered with imperceptible 1 wt.% s-B2P in PLA glass (Supplementary Figs. 79 and 80d). As shown in Fig. 5f, the resultant shells demonstrated yellow-green afterglow after the photo-activation. Therefore, photo-activated UOP PLA glass can act as the decoration of artworks.
Fig. 5: The applications of photo-activated UOP films.
a Overview of writing, reading and erasing procedure of 1 wt.% r-MOP in PMMA film. b Different phosphorescent images written by photo-activation successively onto the same substrate of 1 wt.% r-MOP in PMMA film and erased by leaving in dark for 50 min. c Multicolor phosphorescence images taken after photo-activated by 302 nm based on PMMA films doped different guests. d High-level data encryption based on 1 wt.% r-MOP in PMMA film with different photo-activation time. e Photo-activated UOP films for anti-counterfeiting based on 1 wt.% MOPY in PVAc film. f Photographs of the shells covered with 1 wt.% s-B2P in PLA glass taken under room light and 365-nm lamp off after the photo-activation. Scale bar: 0.5 cm.