
A three-year-old male presented to the pediatric emergency department with headaches and vomiting. He appeared in poor general health, with reduced nutritional status and impaired vigilance. Additionally, the patient exhibited stereotypical respiratory sounds, retching and twitching with hypersalivation. Pupils were normal in size and reactivity. The patient’s medical history, including developmental milestones, was unremarkable and there was no known family history of Rhabdoid Tumor Predisposition Syndrome (RTPS). Cranial MRI-imaging revealed a tumor in the right side of the cerebellum.
Due to the unfavorable anatomical location with proximity to the brainstem and cranial nerves, only a subtotal tumor resection with a macroscopic tumor residue was performed six days after initial presentation. A Rickham reservoir was placed for the purpose of intraventricular chemotherapy and CSF analysis of methotrexate levels as well as cytology.
Results of histopathological analysis revealed a heterogenous malignant neoplasia with embryonal characteristics and immunohistochemical loss of nuclear SMARCB1/INI1 expression, while SMARCA4/BRG1 staining was retained. Ultimately, based on methylation analyses using the Infinium MethylationEPIC BeadChip assay of Illumina, the tumor was classified as ATRT-TYR according to the Heidelberg Brain Tumor Classifier (v12.8). CNV profile in this case was without any aberrations as typical for ATRT, subgroup TYR (data not shown).
Next generation sequencing (NGS) using the TrueSight Oncology 500 assay provided by Illumina identified a variant in the SMARCB1gene: c.1175del; p.(Pro392Argfs*100), with a variant allele frequency (VAF) of 88%. Unlike typical nonsense variants that lead to early termination of protein biosynthesis, this frameshift mutation results in a prolonged protein product with an altered C-terminal.
This specific variant is not listed in databases such as HGMD professional, LOVD and gnomAD, although it has been reported once in ClinVar. According to ACMG guidelines, the variant is classified as likely pathogenic in class four with a tendency toward class five [10]. Additionally, OncoScan analysis of the tumoral DNA identified a near-complete copy number neutral loss of heterozygosity (CNN-LOH) of chromosome 22 (chr22:18,319,179–51,213,826), including the SMARCB1 gene. The CNN-LOH aligns with the high allele frequency of the SMARCB1 variant and provides an explanation for the complete loss of SMARCB1/INI1 expression.
NGS also detected a heterozygous missense variant in SMARCA4 (c.3484G > A, p.Gly1162Ser) in the tumor DNA with a VAF of 56%, affecting the C-terminal helicase domain of the protein. This variant is also not listed in the aforementioned databases. In silico prediction programs (FathmmRank, MetaLrRank, MetaRnnRank, MetaSvmRank) consistently predicted a disease-causing, deleterious effect of the alteration. However, due to insufficient data, it remains uncertain whether this SMARCA4 alteration contributes to the clinical manifestation. According to ACMG guidelines, it is categorized as a variant of uncertain significance [10]. We did not identify a second alteration of SMARCA4 in the tumor DNA by NGS or OncoScan analysis. Thus, it is unlikely that the SMARCA4 variant is causal to disease initiation but it may potentially act as a modifier of disease course. This is backed further by functional studies: The p.G1162S variant lies within the ATPase domain, which is vital for the catalytic activity of the SMARCA4 protein. Variants in this region have been shown to impair SWI/SNF complex activity and contribute to oncogenesis in multiple tumor types [11]. Table 1 provides a detailed overview of the alterations in SMARCB1 and SMARCA4 identified in the tumor tissue.
Table 1 Table of genetic Alterations in SMARCB1 and SMARCA4 identified in tumor tissue
To assess potential germline alterations, NGS and Multiplex-Ligation-dependent Probe Amplification analysis of blood-derived DNA were performed in accordance with the German Genetic Diagnostics Act. Finally, using recently developed approaches no evidence of low-level mosaicism in SMARCB1 or SMARCA4 were detected [12].These findings effectively rule out RTPS and demonstrate that the described alterations in SMARCB1 and SMARCA4 are of somatic origin (Fig. 1a).
Fig. 1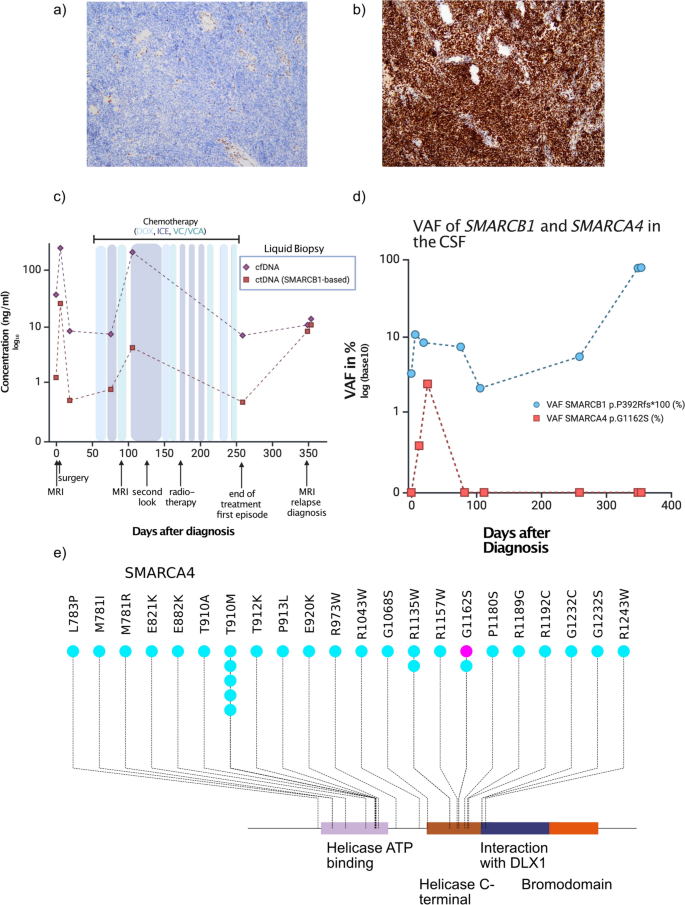
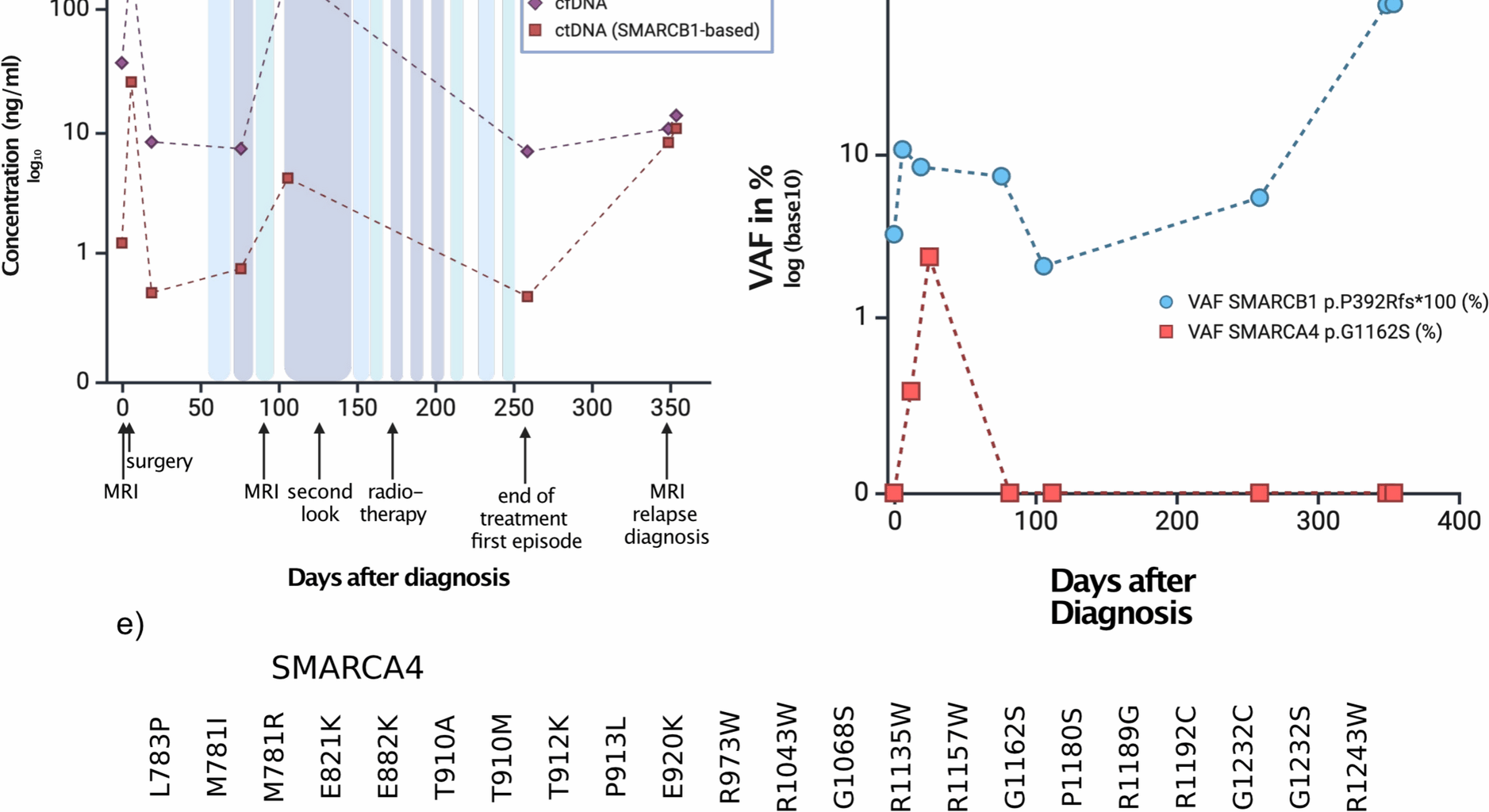
a Immunohistochemical staining of INI-1 in the patient case, which is lost. Panel b shows retained SMARCA4 staining of the same case c cfDNA (violet line) and ctDNA (red line) burden in the time course of the patient. d VAF in the course of time. e Lollipop plot displaying the somatic mutations of SAMRCA4 in medulloblastomas (light blue) as a reference cohort from [14] and the here described case (violet). Displayed are only the Helicase ATP binding domain, the C terminal helicase domain and the Bromodomain as the only protein domains where mutations map to.
To track disease burden based on the known SMARCB1 and SMARCA4 alterations, we included the patient in a liquid biopsy program within the frame of the BZKF (Bayerisches Zentrum für Krebsforschung) which employs isolation of cell free DNA from cerebrospinal fluid, followed by sequencing using a panel of genes, specific for pediatric neurooncology [13]. Here, we detected both the SMARCB1 frameshift variant and the SMARCA4 missense variant at a VAF varying from 2.1 to 10.7% in SMARCB1 and 0 to 2.4% in SMARCA4 depending on time (Fig. 1b).
Following surgery, the patient was enrolled in the SIOPE ATRT01 trial, receiving a total of 12 courses of chemotherapy. The chemotherapeutic treatment regimen consisted of approximately biweekly alternating administrations of doxorubicin (DOX), ifosfamide, carboplatin and etoposide (ICE) and vincristine, cyclophosphamide and actinomycin D (VCA). During the second course of VCA, actinomycin D was omitted due to the initiation of radiotherapy. Intraventricular administration of methotrexate (MTX) was planned in parallel to each course in an age-dependent fashion; however, it was ultimately administered only once via lumbar puncture during the third course of chemotherapy, first due to a cerebrospinal fistula and subsequently due to the start of radiotherapy.
To evaluate the therapeutic response, an MRI was conducted 82 days after the initial presentation following the third course of therapy. Imaging revealed remnants of the tumor around the medulla oblongata, which were initially classified as stable disease but were later reassessed as progressive disease by reference neuroradiology. Using liquid biopsy as a complementary method to MRI monitoring, we confirmed an increase in tumor burden as shown by the elevated levels of cell free DNA (cfDNA) in the CSF, but also in the ctDNA as measured by the VAF of the SMARCB1 variant (Fig. 1c, d). Given that the patient did not display an infection at this time, we assumed that this increase reflected submicroscopic tumor burden.
Based on these findings and the general recommendation to minimize residual tumor via second look surgery, a re-resection of infratentorial tumor parts was performed 3.5 months after first presentation and four courses of chemotherapy, resulting in the removal of the largest tumor mass, with only minimal residues in direct proximity to the medulla oblongata remaining in situ.
The patient subsequently underwent proton beam therapy and received the remaining chemotherapy, which was administered partially concurrently, with close disease monitoring throughout the treatment period. His treatment was completed according to protocol. After approximately nine months, at the end of therapy, an MRI scan revealed stable disease without suspicion of relapse. However, liquid biopsy monitoring at that time showed an increase of the VAF of the SMARCB1 variant (Fig. 1c) while total cfDNA concentration remained low. This increase proved to be an early indicator of disease activity.
Heterozygous missThe hitherto identified SMARCA4 variants in ATRT are exclusively homozygous and inactivating. The SMARCA4 variant described here (p.G1162S) is localized in the helicase C-terminal domain of the protein. To contextualize the variant, we reviewed the mutational data from a large-scale study on medulloblastoma [14] which identified multiple variants within the C-terminal helicase domain, including one tumor harboring the p.G1162S substitution (Fig. 1e). Of note, the same heterozygous SMARCA4 mutation has also been detected in adult T-ALL relapses (Sentis et al. Genome Biology 2020).
Three months later, follow-up imaging revealed metastatic spread, and the patient is currently undergoing treatment for early recurrence of his AT/RT. This was paralleled by an increase both of the total cfDNA but also in the VAF of the SMARCB1 gene alteration.