
Participants from the 2000HIV study
The 2000HIV study is a prospective multicentric observational longitudinal cohort of virally suppressed PLHIV15. Participants were recruited from October 2019 to October 2021. The cohort included both discovery and validation cohorts. Participants in the discovery cohort were recruited from three specialized Dutch HIV treatment facilities, two university medical centers and one large general hospital (Radboudumc Nijmegen, Erasmus MC Rotterdam and OLVG Amsterdam). Participants in the validation cohort were recruited at a different medical facility: a large general hospital (Elisabeth-TweeSteden Ziekenhuis Tilburg). Inclusion criteria included HIV-1 infection, age 18 years or older, 6 months of ART and a most recent HIV-1 RNA level of less than 200 copies per milliliter. Individuals with spontaneous HIV-1 control without ART could participate if viral loads were less than 10,000 copies per milliliter for at least 5 years and CD4+ T cell counts were stable (>500 cells per mm3). Exclusion criteria included no informed consent, insufficient communication due to language barriers or other issues, current pregnancy, detectable viral hepatitis B or C DNA by polymerase chain reaction or signs of any current acute infection. Extreme clinical phenotypes, such as spontaneous ‘elite’ controllers, and rapid progressors were identified as described previously15 as well as carotid plaque assessment using B mode ultrasound.
The 2000HIV study was approved by Independent Review Board Nijmegen (NL68056.091.81) and published at ClinicalTrials.gov (NCT03994835). Written informed consent was received from participants before inclusion in the study. All experiments with human samples were conducted following the principles of the Declaration of Helsinki.
Multi-omics measurementsGenomics—genotyping array
DNA was extracted from each participant’s whole blood. The Illumina Infinium Global Screening Array was used for genotyping all participants of multiple ethnicities in the 2000HIV cohort. PLINK version 1.90b44 was used to perform quality control on raw variants and samples before imputation. The dataset excluded genetic variants with call rate genotype missingness of more than 5% and deviations from Hardy–Weinberg equilibrium (HWE) with P < 10−6. The HWE exact test was performed with variants stratified by ethnicity. We excluded samples with a call rate of less than 97.5% and heterozygosity rates that deviated more than 3 s.d. from the mean rate per self-reported ethnicity. Genetic variants that passed quality control were converted from GRCh37 to GRCh38 using the UCSC liftOver tool45. Next, TOPMed Freeze 5 was used on genome build GRCh38 to align strands to the TOPMed reference panel. We used the McCarthy group tools for alignment (https://www.well.ox.ac.uk/~wrayner/tools/). After quality control, 582,404 variants from 1,864 individuals were kept for the imputation process. The filtered raw variants were uploaded to the TOPMed Imputation server and compared to the TOPMed (version r2 on GRCh38) reference panel. The imputed variants were filtered using BCFtools stratified by ethnicity46, excluding variants with low imputation quality scores (R2 < 0.3 or empirical R2 < 0.7) or minor allele frequency (MAF) < 1%. This yielded 10,810,841 variants from 1,864 members of the 2000HIV multi-ancestry cohort.
The above-mentioned quality control and imputation procedure was applied independently to the European ancestry discovery and validation cohorts. During quality control per marker, variants with a call rate greater than 5%, MAF < 1% and deviation from HWE (P < 10−6) were removed from the European datasets of the discovery and validation cohorts. Samples with a call rate less than 97.5%, heterozygosity rates that deviated over 3 s.d. from the mean and ethnic outliers identified through principal component analysis (PCA) were removed during quality control. Individuals were defined as ethnic outliers if their genetic principal component 1 (PC1) and/or principal component 2 (PC2) deviated by more than 3 s.d. from the mean PC1 and/or PC2 of the European population from the 1000 Genomes Project47. After quality control and imputation, the imputed variants were filtered as described above, yielding 9,148,674 and 9,130,602 SNPs from 1,003 and 257 individuals in the discovery and validation cohorts, respectively.
After imputation, PLINK 2.0 (ref. 48) was used to perform quality control. Any variants failing the HWE test at P < 1 × 10−12 and those with MAF < 1% and R2 < 0.05 were eliminated. In total, 8,944,122 imputed SNP variants were kept for further analysis after quality control.
Epigenomics—methylation array
A total of 1,914 samples underwent DNA methylation. The Radboudumc Genetics Department isolated DNA from EDTA whole blood using the chemagic STAR automated configuration (consisting of the Microlab STAR and Chemagen Magnetic Separation Module 1; Hamilton Robotics) combined with Chemagen nucleic acid extraction technology with magnetic polyvinyl alcohol (M-PVA) beads, which follows a standard and automated bind–wash–elute protocol. A NanoDrop spectrophotometer was used to determine the DNA concentration and 260/280-nm ratio. Samples were then normalized to 50 ng µl−1 in TE buffer and randomly distributed among plates. The Illumina Infinium MethylationEPIC BeadChip array was used to profile DNA methylation across the genome. Standard sample-based and probe-based quality control was carried out. DNA methylation values were estimated from raw IDAT files using R’s ‘minfi’ package (version 4.2.0). Preprocessing steps eliminated two sex mismatch samples from the discovery cohort and one low-quality sample from the validation cohort (call rate <99%). Probes (discovery: n = 2,743 and validation: n = 2,641) with missing methylation values (detection P > 0.01) in more than 10% of samples, as well as probes on the sex chromosome (n = 19,627), were excluded from the downstream analysis. Because the participants are European, we also removed probes containing SNPs at target CpG sites with MAF > 5% in European populations as well as probes that mapped to multiple loci (both discovery and validation: n = 52,173). Next, we applied stratified quantile normalization. Methylation β-values were calculated as a percentage: β = M / (M + U + 100), where M and U represent methylated and unmethylated signal intensities, respectively. The β-values were then transformed to M-values as log2(β / (1 − β)), and M-values were used in all subsequent analyses.
Transcriptomics—RNA sequencing
For transcriptomics analysis, PBMCs were sequenced in bulk using short-read sequencing with current Illumina technology (>30 million reads per sample). STAR alignment was used to map the sequencing reads to the most recent version of the human reference genome NCBI build 38. Gene expression was estimated using the HTSeqCount function from DESeq2 with the most recent Ensembl gene annotation. The DESeq2 pipeline is used to process raw counts by applying rlog transformation, normalization and exclusion of low abundant transcripts. Further details on the quality control of the transcriptomics data can be found in the paper describing the cohort15.
Proteomics—Olink platform
Circulating plasma protein expression was measured using a commercially available multiplex proximity extension assay (PEA) from Olink Proteomics AB in three batches. This study used the entire library (Olink Explore 3072), which included 3,072 targeted proteins organized into eight 384-plex panels focusing on inflammatory, oncological, cardiometabolic and neurological proteins. Protein measurements were delivered as normalized protein expression (NPX) values following a quality control and normalization process developed and provided by Olink. NPX values are derived by subtracting the extension control and the plate values from Cq values. A correction factor is applied to shift the scale, and all values are reported in the log2scale. Bridging normalization was used to remove batch effects in each of the eight panels from the Olink Explore 3072 platform, and IL-6, TNF, CXCL8, LMOD1, SCRIB and IDO1 were measured as technical duplicates for quality control purposes. Strong correlations were observed between the technical duplicates among panels, and, therefore, we selected the measurements from the inflammatory panel. Next, we excluded proteins with limit of detection (LOD) ≥ 25 of the samples (n = 547 proteins were excluded), resulting in 2,367 proteins for follow-up analysis. Next, during quality control per sample, we performed PCA using the NPX. Outliers were defined as those samples falling above or below 4 s.d. from the mean of PC1 and/or PC2. In total, seven samples were excluded based on PCA, resulting in 1,910 samples analyzed. After this, samples from individuals of European genetic ancestry were selected for this analysis.
Metabolomics—mass spectrometry
The abundance of 1,720 circulated metabolites in 1,902 serum samples was determined using General Metabolicsʼ untargeted metabolic platform. Untargeted metabolome profiling was carried out on plasma samples using flow injection electrospray time-of-flight mass spectrometry, as described previously, in collaboration with General Metabolics49. Metabolites were identified based on the mass-to-charge ratio (ion m/z). Prior to analysis, the raw metabolome data were averaged and normalized to remove duplicate peak intensity using a moving median normalization. PCA was then used to identify potential outlier samples. Metabolites were annotated and classified according to the metabolomic source (endogenous, food or drug) and chemical taxonomy using publicly available data from the Human Metabolome Database (HMDB)50. According to the HMDB, 851 of the 1,720 metabolites were identified as endogenous and were used for further analysis.
Cytokine production assay
PBMCs were stimulated with a range of whole (inactivated) pathogens, pattern recognition receptor ligands, other pathogen-derived antigens and viral stimuli to quantify the capacity for cytokine production. Round-bottom 96-well plates (Greiner Bio-One) containing 500,000 cells per well were used for the stimulations, which were carried out for either 24 hours at 37 °C and 5% CO2 or 7 days (with 10% human pool serum added). Supernatants were gathered and kept at −20 °C until ELISA was used to measure the relevant cytokines. Specifically for the 24-hour stimulations, the stimulants used included poly I:C, LPS, imiquimod, IL-1a, HIV viral envelope, CMV and S. pneumoniae, and the cytokines measured included IL-1β, IL-1Ra, IL-6, IL-8, IL-10, MCP-1, MIP-1a and TNF. For the 7-day stimulations, the stimulants used included E. coli, S. aureus, S. pneumoniae, M. tuberculosis, C. albicans (conidia), C. albicans (hyphae) and PHA, and the cytokines measured included IL-5, IL-10, IL-17, IL-22 and IFNγ. A detailed explanation of the concentrations, manufacturers and strains can be found in the cohort publication.
Data processing was as follows. For the 24-hour experiment, samples from 1,742 participants were measured, of which 42 samples were excluded for being RPMI positive, defined as having concentrations of above 2× the lower limit of detection (LLOD) after RPMI stimulation in two out of TNF, IL-1b or IL-6. Outliers on PCA were defined as those with ±4 s.d. away from the mean of PC1 and/or PC2 (n = 13). Data from 1,687 participants were used in downstream analysis. For the 7-day experiment, samples from 1,744 participants were measured, of which 42 samples were excluded for being RPMI positive, and 20 outliers on PCA were removed as in the 24-hour experiments. Data from a total of 1,682 participants were used in downstream analysis. For the current analyses, only data from participants of European genetic ancestry were used.
Cell count
Whole blood samples were immunophenotyped using three flow cytometry panels, each with 17–20 markers, and custom-made tubes containing dry antibodies from DURA Innovations Technology (Beckman Coulter). Cells were collected in a 21-color, six-laser CytoFLEX-LX (Beckman Coulter) with CytExpert software 2.3. Daily instrument quality control and standardization were carried out with CytoFLEX Daily QC Fluorospheres (Beckman Coulter, cat. no. B53230), CytoFLEX Daily IR QC Fluorosphere Beads (Beckman Coulter, cat. no. C06147) and SPHERO Rainbow Calibration Particles, 6 peaks (Spherotech, cat. no. RCP-30-5A-6). The data analysis was performed using Kaluza version 2.1.2 and Cytobank Platform version 9.0 (Beckman Coulter).
Multi-omics integration
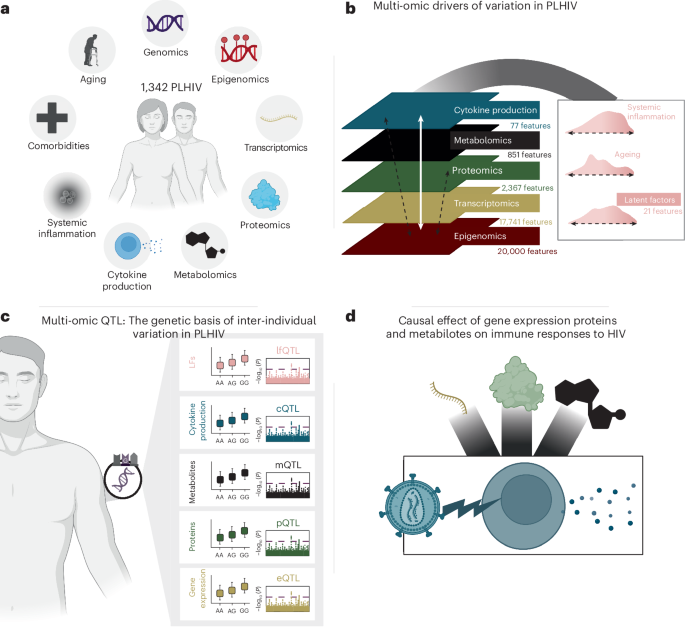
All data modalities except genomics, namely epigenomics, transcriptomics, proteomics, metabolomics and cytokine production, were integrated by applying MOFA14. Each modality was preprocessed independently, and transcriptomics, metabolomics and cytokine production data were log normalized. To reduce the number of features in the epigenomics data (DNA methylation), the top 20,000 CpG probes with higher variance were selected. Each modality was corrected for age, sex and the institute in which the samples were collected by extracting the residuals of a linear model. Only samples with all modalities available in the discovery cohort were included. MOFA was run by applying the R function ‘run_mofa’ with default parameters, 30 factors and view scaling. Factors that correlated with the average of all features in any of the modalities with an absolute correlation above 0.6 were considered technical artifacts and were discarded for interpretation. Only factors that explained more than 1% of the variance across all modalities were considered for interpretation. This resulted in a total of 21 LFs.
Association between LFs and other variables
The 21 LFs identified by running MOFA were tested for association with the immunological data, covariates and clinical variables present in the study.
To associate with IL-1β levels, normalized and scaled protein, gene expression and cytokine values were used. A cytokine ‘IL-1β score’ was calculated by averaging IL-1β production among all 24-hour stimulations. Each of these three values was correlated to each of the 21 LFs using Pearson’s correlation, and the significance of the correlation was assessed by applying FDR correction. The inter-correlation between IL-1β scores was also calculated using Pearson’s correlation (Extended Data Fig. 8), including a measure of gene expression to protein levels ratio by calculating the difference between the normalized IL-1β levels between both.
A list of variables was assessed to associate LFs with covariates and clinical variables (Supplementary Data 2 and 3). Binary variables—for example, biological sex—were tested using the Wilcoxon rank-sum test. Categorical variables with more than two categories were tested by applying the Kruskal–Wallis test. Continuous variables were tested using Pearson’s correlation. All P values resulting from the multiple tests were corrected by applying FDR correction. The sign of the association was defined differently depending on the variable type: for binary variables, it was defined as the difference in the median between groups; for continuous variables, as the correlation sign; and for variables with multiple categories it was left as positive for visualization purposes.
Molecular profiling of the multi-omic LFs
For each of the LFs, a ‘molecular profile’ was constructed to interpret the biological functions that it is capturing.
A set of features with significant weights was extracted for each factor and data modality. These significant weights were defined by standardizing the feature weights per factor and extracting features whose weight corresponded to a probability of 1% of having more extreme values, assuming a standard normal distribution.
Factors were tested for enrichment in different categories by running MOFA2’s R function ‘run_enrichment’, which is based on the principal component gene set enrichment method. Methylation, gene expression and protein weights were tested for enrichment against the gene sets defined by blood transcriptome modules51. Cytokine profile measures were tested for enrichment based on groupings made at the time of stimulation, the stimulant applied and the cytokine measured. All enrichments were run per sign—that is, separately for features with positive and negative weights—and with a minimum size of the feature set of 5.
Multi-omic factor validation
All LFs estimated by running MOFA in the discovery cohort were interpolated to the validation cohort. All data were normalized following the same steps as in the discovery cohort except for the correction for the institute of collection, as the validation cohort only includes samples that were collected in a different institute. Features with significant weights, as defined in the ‘Molecular profiling of the multi-omic LFs’ subsection, were extracted and used for the validation cohort, as this may reduce data overfitting. Factors were calculated by performing matrix multiplication between the preprocessed data and the significant weights. To test for the robustness of this method, the same approach was performed on the discovery cohort showing sufficient correlation (Extended Data Fig. 9).
QTL mapping
Using both the discovery and validation cohorts, we performed QTL mapping for gene expression (ndisc = 1,048, nval = 260), protein levels (ndisc = 1,064, nval = 266) and metabolite levels (ndisc = 1,069, nval = 267). For cytokine response QTL, each cytokine stimulation pair encompassed a different number of samples, ranging from 196 to 1,031 for the discovery cohort and from 41 to 260 for the validation cohort. We mapped the inverse-rank transformed values baseline omics (gene expression, proteins and metabolites) as well as cytokine response to the genotype data using a linear model that included age, sex, body mass index (BMI), seasonality, inclusion before the COVID pandemic, COVID vaccination and recruitment center. A sine and cosine wave with a period of 365.25 days was used to model seasonality. When combined, these two terms can create a sine wave with any phase and a yearly frequency52. QTL mapping was performed using the MatrixEQTL package53. Co-localization analyses were performed using the coloc R package54. We calculated study-wide P values by dividing GWs (5 × 10−8) by the number of effective tests. The number of effective tests was calculated following the formula described by Li et al.55, which takes into account the correlation matrix of the measurements.
For the comparison of the eQTL, pQTL and mQTL in the discovery cohort of PLHIV as opposed to healthy people, we used the public QTL databases GTEx21 and UKB-PPP22 as well as the mQTL from a healthy cohort23. We took the lead SNPs for all the GWs loci and looked for the same combination of SNP to trait in the corresponding full summary statistics, regardless of the significance of the association in the healthy cohort. We then estimated the concordance by considering the direction of effects as well as the correlation of the effects.
Mendelian randomization
A systematic Mendelian randomization analysis was done using the R package TwoSampleMR56, with the inverse-variance weighted (IVW) method57 being the primary approach. For each exposure (gene expression, protein levels and metabolite levels), genetic instruments were carefully selected to ensure adherence to the assumptions. First, they were strongly associated (P < 1 × 10−5) with the exposure in the discovery cohort. They were then further validated, excluding any SNPs not nominal significant in the validation cohort. SNPs with MAF < 0.05 were filtered out. Lastly, SNPs associated with more than four other traits within the same exposure were considered too pleiotropic and were excluded. Next, stringent clumping was performed (r2 = 0.001, kb = 10,000) to ensure independent SNPs. Only if there were still at least three SNPs remaining, Mendelian randomization was performed. For Mendelian randomization with significant IVW (P < 0.05), sensitivity analyses were performed—horizontal pleiotropy, heterogeneity and leave-one-out. We performed the same Mendelian randomization in the other directions (bidirectional), and any results that had a nominal significant IVW result in the other direction, while also passing all the sensitivity checks, were excluded.
Inflammasome score calculation
To estimate the activity of the inflammasome complex at both transcriptional and proteomic levels, a score was calculated per sample. For each sample, the inflammasome score was defined as the average of expression of all genes in the gene set using the covariate corrected values. The following gene sets were extracted from the Molecular Signatures Database (https://www.gsea-msigdb.org/gsea):
GOBP_POSITIVE_REGULATION_OF_INFLAMMASOME_MEDIATED_SIGNALING_PATHWAY.v2023.2.Hs
REACTOME_INFLAMMASOMES.v2023.2.Hs
GOCC_CANONICAL_INFLAMMASOME_COMPLEX.v2023.2.Hs
REACTOME_THE_NLRP3_INFLAMMASOME.v2023.2.Hs
GOCC_NLRP3_INFLAMMASOME_COMPLEX.v2023.2.Hs
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.