
Ethics approval
All patients provided written informed consent before enrollment. This prospective clinical trial was compliant with the Health Insurance Portability and Accountability Act and the design was preregistered at https://classic.clinicaltrials.gov/ct2/show/NCT04949776. In March 2021, the study protocol and the informed consent received a favorable ruling from the Institutional Review Board (IRB) at Reina Sofía University Hospital of Córdoba Research Ethics Committee (IRB No 4932).
Clinical trial design and patients
This is a prospective paired noninferiority experimental study.
The individual deidentified participant dataset, data dictionary defining each field and study protocol are publicly accessible via Zenodo at https://doi.org/10.5281/zenodo.17625633 (ref. 24).
This clinical trial was carried out in the Córdoba Breast Cancer Screening Unit, Córdoba, Spain, as part of the Andalusian screening program in Spain. Women participants aged 50 to 71 years old (including women who reach that age in the year of appointment) are invited to participate in the screening program at 2-year intervals (ClinicalTrials.gov ID: NTC04949776).
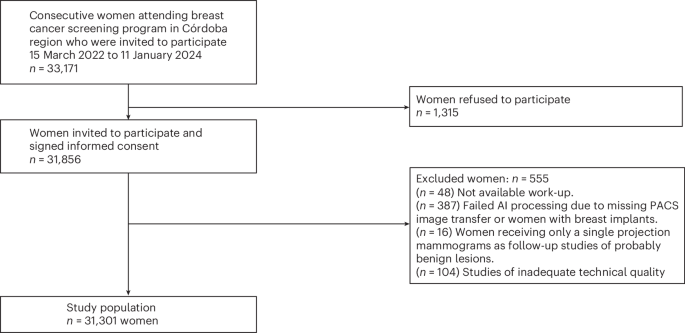
All women participating in the screening program from 15 March 2022 to 11 January 2024 were eligible and invited to participate in this clinical trial. They were asked to sign an informed consent form. For screening purposes, sex and age data were defined by reference to the population census. All women aged 50 to 71 years, regardless of socioeconomic status, were considered.
After signing informed consent, women were excluded from the clinical trial if they had symptoms or signs of suspected breast cancer; if they had breast prostheses; or if their images could not be processed by the AI system (for example, because of the presence of visible breast implants or because of erroneous PACS image transfer).
To investigate the hypothesis and to account for ethical aspects, this clinical trial had a paired design (Fig. 2). Each participant received both reading strategies:
(1)
The standard of care: double human reading without AI support.
(2)
The AI strategy: double human reading with AI support (by two additional radiologists) only for cases classified by the AI system with a score of 8 to 10 (approximately 30% of the studies most likely to have cancer). Cases classified by the AI system with a score of 1 to 7 were considered low risk (approximately 70% of the studies) and automatically classified as normal.
Procedures
Each participant underwent a standard four-view DM or DBT exam based on resource availability, according to the standard of care. There were no rules governing imaging modality selection. The assignment of women to the different types of mammography equipment was not changed for this study. Images were acquired using four devices: three DM devices (Lorad Selenia, Hologic) and one DBT device (3Dimensions, Hologic).
All exams were independently double read without consensus or arbitration. Screening readings were randomly assigned according to availability of the nine dedicated breast radiologists in the hospital’s radiology department (3 to 21 years of experience in breast cancer screening), regardless of whether they were readings with or without AI assistance and regardless of the reader’s experience. The mammogram offered to the reader was the oldest one performed that had not been read by that same reader. In each reading, the radiologist noted the breast density (measured subjectively according to the BI-RADS classification31), the presence of findings and their location in the breast, the assigned BI-RADS category (1 to 5) and the decision whether or not to recall. Women were recalled if any reader decided to recall.
Women received a single result (recall or no recall) once all readings were completed. If recalled, additional exams (special mammography views, tomosynthesis, contrast-enhanced mammography and/or ultrasonography) and, when required, biopsy were performed at the women’s referral unit. The screening program did not include ultrasonography or other additional examinations for women with high breast density. No adverse events were reported in this study.
This clinical trial used a commercially available AI system for breast cancer detection, Transpara (version 1.7 ScreenPoint Medical), which had been used for previous research studies by the same group of radiologists involved in this study.
The AI system’s performance, generalizability and clinical utility has been previously investigated in over 30 other peer-reviewed scientific publications. Most significantly, researchers have shown that this system can achieve stand-alone breast cancer detection performances comparable to radiologists10, that radiologists become more accurate when using such an AI system for concurrent decision support reading mammograms, leading to higher screening CDRs18,19, and that it is safe and effective to implement in breast cancer screening to replace the need for the double reading18,19.
The AI system automatically detects and classifies regions suspicious of breast cancer in radiological images that adhere to the input compatibility criteria of the system and with standard Digital Imaging and Communications in Medicine. This version of the system can be used in combination with DM and DBT images acquired with systems from all major mammography equipment manufacturers (Siemens Healthineers, Hologic, General Electric, Giotto, Planmed, Fujifilm). The system can analyze any number of standard (craniocaudal, mediolateral oblique) and nonstandard (exaggerated, lateral) views per breast. Images of women with breast implants are not compatible unless the implant has been displaced during compression.
The AI system outputs per exam are (1) explainable image overlay markings for regions suspicious of breast cancer per view, alongside a suspiciousness score per region between 1 and 100 and an indication of the finding morphological type, and (2) an exam-based indication of the risk that the exam contains a visible lesion suspicious of breast cancer that requires additional follow-up (three categories: 1–7 low risk, 8–9 intermediate risk, 10 elevated risk).
The system uses deep convolutional neural networks as backbone algorithms to analyze DM and DBT images and detect lesions suspicious for breast cancer. Convolutional neural networks are state-of-the-art machine learning tools for image classification. Different convolutional neural networks are trained with different architectures and training datasets for malignancy detection of different morphological subtypes and taking a variable number of images as input.
The AI system was developed using a large-scale heterogeneous curated database of more than 15 million two-dimensional and three-dimensional X-ray breast images acquired in real-world breast cancer screening programs and diagnostic clinics. The data originated from over 15 sites across 10 countries across North America, Europe and Asia, including over 15,000 malignant cases (pathology proven). Negative cases were confirmed through multiple years of follow-up.
The AI system software version and operating points used in the present evaluation were established before the clinical trial. The choice of a score of 7 as the cutoff point for indicating that human reading was not necessary was based on previous studies conducted by this research team and other published research4,5,25. None of the clinical data used in this clinical trial was used in any aspect of algorithm development.
The human readers in the AI strategy had available the information provided by the AI system (marks according to the type of lesion with a score according to the probability of having cancer and global score of the study), but the radiologist was the person who ultimately made the decision to recall a woman or not.
Data on the procedures, outcomes, participants and detected cancers were retrieved from the medical records. Race and ethnicity were not individually recorded, but the majority of the target population was Caucasian.
Safety
The study protocol was reviewed and approved by the institutional ethics committee, which determined that the study posed minimal risk to participants.
Safety monitoring focused on nonphysical risks potentially associated with the use of AI-based image analysis, including data integrity, patient privacy and the potential for diagnostic error. All mammographic images were fully anonymized prior to analysis, and data handling complied with applicable data protection regulations. Any unexpected incidents related to data processing, algorithm malfunction or breaches of confidentiality were predefined as reportable events and were monitored throughout the study period.
Outcomes
The primary outcomes were workload, CDR and RR calculated globally. Workload was computed as the absolute number of radiologist readings in each strategy. A cancer detected during the diagnostic work-up after a recall and confirmed by pathology was considered the gold standard.
The CDR was estimated as the number of cancers detected per 1,000 screening examinations for each strategy. Cases that were not recalled or that were recalled with malignancy not demonstrated on complementary imaging tests or biopsies were considered negative. Follow-up lasted for a minimum of 180 days after screening mammography. For each case, a strategy was positive if one or both readings included referral.
As secondary objectives, PPV of recalls and FPR were assessed globally. A subgroup analysis by modality was performed and workload, CDR, RR, PPV of recalls and FPR were calculated for DM and for DBT separately. The PPV of recalls was calculated as the number of cancers diagnosed per 100 women recalled for each strategy. The FPV was calculated as the number of women recalled who were not finally diagnosed with cancer for each strategy per 100 negative screening examinations.
Sample size
For the sample size calculation, the method by Connor32 for paired studies was used, taking into account the sample size necessary to demonstrate noninferiority (5% margin) in terms of sensitivity for cancer detection via McNemar’s z-test (one-sided). An alpha (type I error estimate) value of 5% and a beta (type II error estimate) of 20% (representing a power for the study of 80%, 1 − beta) was used. In addition, we used the parameters of an assumed cancer incidence of 6 of 1,000 (value of cancer incidence when AI is not used in screening based on initial experience and more recently verified in ref. 5). We also calculated how many cancers would be detected by the standard strategy alone and how many would be detected by the AI strategy proposed in this study using the database and methods used in ref. 4 but using Transpara software version 1.7.0 (as opposed ref. 4, which used version 1.6.0, which has inferior algorithms). In the results obtained, 1.8% of cancers were detected using the standard strategy but not using the proposed strategy with AI, while the opposite occurred in 9% of cancers. With these parameters, the sample size needed was defined as 27,000 women in order to demonstrate the noninferiority of the AI strategy over the standard strategy globally, including both DM and tomosynthesis studies.
The three coprimary endpoints were CDR, RR and workload. It is worth mentioning that no formal multiplicity correction was applied between them for the sample size calculation. Readers are encouraged to interpret the findings in the context of these coprimary outcomes.
Statistical analyses
The hypothesis raised in this work is that a partially autonomous AI-supported screening strategy (that is, no human reading in cases with low-AI risk (AI strategy)) would allow for reducing the workload of a screening program that uses both DM and DBT exams compared to the standard-of-care reading without demonstrating inferiority in terms of the CDR and RR of the program. Descriptive analyses of the variables considered in the study were carried out in the complete dataset of women. Categorical variables were expressed as absolute and relative frequencies, while continuous variables were expressed as median and interquartile range. To compare the AI-assisted strategy with the standard-of-care strategy, we evaluated the CDR and RR, as well as secondary metrics including the PPV of recall and FPR. For all variables, percentages were calculated for each strategy, along with corresponding 95% CI. Differences between AI and standard strategies were assessed using Wald statistics with or without the Bonett–Laplace adjustment33, as appropriate.
For the primary outcomes (CDR and RR), we conducted noninferiority testing using the following approach: reference values for the CDR and RR were identified from previously published noninferiority studies and screening benchmarks in the literature. Based on clinical consensus, a 5% relative tolerance margin over those values was considered acceptable for declaring noninferiority. For the CDR (where higher values are better), a noninferiority margin was defined as a 5% relative reduction from the reference value. For the RR (where lower values are generally preferred), the noninferiority margin was set as a 5% relative increase from the reference value.
These thresholds were used as the minimum acceptable performance levels for the AI-assisted strategy. One-sided 97.5% CIs were established for the difference in proportions (AI standard) for the CDR and RR. These were derived from two-sided 95% CIs. Noninferiority was declared if the lower limit of the one-sided CI exceeded the predefined noninferiority margin. Visual representation of these thresholds is shown in Extended Data Figs. 1 and 2.
If noninferiority was demonstrated for a given outcome, we proceeded to test for superiority using a paired exact McNemar test. For PPV and FPR, noninferiority was not formally tested. Instead, standard two-sided hypothesis tests were conducted: a z-test was used for comparing PPV (assuming independent data) and a paired McNemar test was used for comparing FPR.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.