
Chemicals
Unless otherwise indicated, all chemicals and reagents used in this study were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Porcine oocyte collection and in vitro maturation
Porcine ovaries (mixed Yorkshire, approximately 6-month-old gilts) were obtained from a local slaughterhouse (Dong-A Food, Republic of Korea) and transported to the laboratory within 2 h in physiological saline at 37–39 °C. After washing the ovaries twice with physiological saline, cumulus–oocyte complexes (COCs) were aspirated from the antral follicles (3–6 mm) using an 18 G needle attached to a 10 mL disposable syringe and then collected in a 15 mL conical centrifuge tube.
After 10 min settling at 37 °C, the supernatant was removed, and the precipitate was resuspended in HEPES-buffered Tyrode’s medium containing 0.05% (w/v) polyvinyl alcohol (TLH-PVA). COCs with more than three layers of compact cumulus cells and a homogenous cytoplasm were selected for in vitro maturation. Groups of 60 randomly selected COCs were transferred into each well of a four-well plate containing 480 µL of maturation medium comprising TCM199 (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 0.6 mM cysteine, 0.91 mM sodium pyruvate, 10 ng/mL epidermal growth factor, 75 µg/mL kanamycin, 1 µg/mL insulin, and 10% (v/v) porcine follicular fluid. Thereafter, the COCs were incubated for 40–42 h at 39 °C in an atmosphere of 5% CO2 with humidified air. For the first 22 h, the COCs were cultured in maturation medium containing hormones (10 IU/mL equine chorionic gonadotropin and 10 IU/mL human chorionic gonadotropin; Daesung Microbiological Labs, Uiwang, Korea). After this period, the hormones were removed, and the COCs were cultured for an additional 18–20 h in hormone-free maturation medium. Next, COCs were denuded using 0.1% hyaluronidase by gentle pipetting and washed thrice in TLH-PVA medium. Oocytes with visible first polar bodies and a uniform ooplasm were selected for embryo production.
Generation of porcine embryos
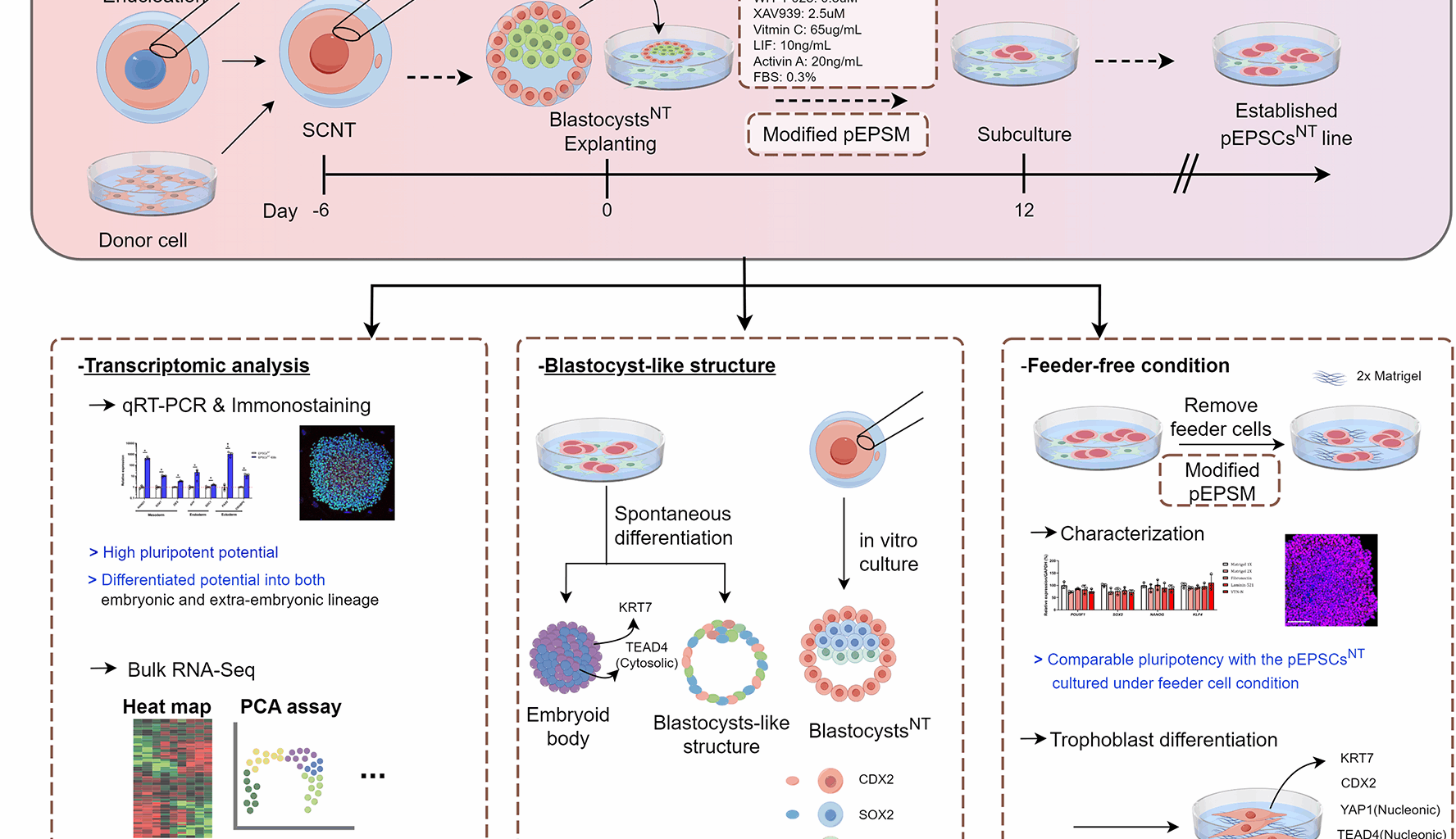
To establish pEPSCs, IVF, PA, and SCNT were performed as previously described [47, 48, 49]. Briefly, weekly shipped liquid semen (Darby Genetics Inc., Gyeonggi-do, Republic of Korea) was used for IVF. After washing twice with Dulbecco’s phosphate-buffered saline (dPBS) containing 0.1% bovine serum albumin (BSA) via centrifugation, sperm pellets were resuspended in modified Tris-buffered medium (mTBM) [50]. Fifteen mature oocytes were placed into each mTBM droplet (40 µL), followed by sperm introduction to achieve a final concentration of 5 × 105 sperm/mL. After 20 min of co-incubation, loosely attached sperm cells were carefully removed from the zona pellucida of the oocytes using precise pipetting techniques. After washing twice, the oocytes were incubated in fresh mTBM for 5–6 h.
For PA, mature oocytes were initially placed in calcium-free HEPES-buffer Tyrode’s medium supplemented with 0.2% BSA (TLH-BSA). Subsequently, the mature oocytes were rinsed twice with an activation solution (pH: 7.0–7.4; osmolarity: 280 mOsm/L; composition: 280 mM mannitol, 0.001 mM CaCl2•2H2O, and 0.05 mM MgCl2•6H2O). Subsequently, the oocytes were placed between two electrodes in a chamber overlaid with activation solution and subjected to two direct-current pulses of 120 V/mm for 60 µs using a LF101 Electro Cell Fusion Generator (Nepa Gene Co., Ltd. Chiba, Japan). These activated oocytes were immediately transferred into porcine zygotic medium-3 (PZM-3) [51] supplemented with 7.5 µg/mL cytochalasin B (CB; C6762) for 3 h of incubation.
For SCNT, mature oocytes were incubated for 5 min in TLH-BSA medium supplemented with 5 µg/mL CB and 5 µg/mL Hoechst 33342 (B2261). After washing several times, oocytes were placed into a drop of TLH-BSA with 5 µg/mL CB, after which they were enucleated by aspirating the polar body and adjacent metaphase II spindle-containing ooplasm with a glasses pipette (16 μm diameter) (Humagen, Charlottesville, VA, USA). After trypsinization, donor cells with smooth surfaces were selected for transfer into the perivitelline space of the enucleated oocytes using a fine injection pipette. The couplets were rinsed twice with activation solution, placed between two electrodes in a chamber overlaid with fusion solution (pH: 7.0–7.4; osmolarity: 260 mOsm/L; composition: 260 mM mannitol, 0.1 mM CaCl2•2H2O and 0.05 mM MgCl2•6H2O), and subjected to direct-current pulses of 160 V/mm for 60 µs using a LF101 Electro Cell Fusion Generator. After 30 min, the membrane fusion status of oocytes in the TLH-BSA solution was confirmed by stereomicroscopy. Fully fused oocytes were transferred to PZM-3 medium, which was supplemented with 2 mM 6-dimethylaminopurine and 0.4 mg/mL demecolcine for 4 h of incubation.
Embryos obtained via IVF, PA, and SCNT were then transferred into 30 µL PZM-3 droplets (10 gametes/drop) covered with pre-warmed mineral oil. Thereafter, the embryos were incubated at 39 °C in a humidified atmosphere comprising 5% O2, 5% CO2, and 90% N2 for a developmental period of 6 or 7 days. The PZM-3 droplets were replaced on days 2 and 4 of culture.
Cultivation of donor cells for SCNT
Porcine fetal fibroblasts derived from a fetus (Landrace × Duroc crossbreed) on embryonic day 40 were prepared as donor cells following the methodology outlined in previous studies [52, 53]. Before SCNT, the cells, which were stored in liquid nitrogen (− 150 °C), were thawed and cultured in cell culture medium composed of Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS), 1× non-essential amino acids (NEAA), 1× glutamine, 1× ß-mercaptoethanol, and 1× antibiotic–antimycotic (Anti-anti) at 37 °C under 5% CO2 in air. After cultivating for 3–4 days to achieve approximately 100% confluence, the donor cells were subjected to trypsinization for 1 min to dissociate the monolayer into single cells for subsequent use in nuclear transfer. All media and reagents used in this culture were purchased from Gibco.
Feeder cell preparation
Mouse embryonic fibroblast (MEFs) derived from embryonic day-13.5 ICR mouse, purchased from DBL (Seoul, Republic of Korea), were utilized as feeder cells. The mouse was euthanized by cervical dislocation, and all efforts were made to minimize suffering. The preparation procedure involved removing the fetal heads, internal organs, and limbs. The remaining tissues were minced, washed in dPBS, and centrifuged at 2,000 rpm for 3 min at least twice. Thereafter, the MEFs were cultured in the aforementioned cell culture medium and maintained at 37 °C under 5% CO2 in air. To inactivate the MEFs, they were treated with 10 µg/mL mitomycin C (Roche, Basel, Switzerland) at passage 1–2 for 2 h. Subsequently, the cells were plated at a density of 5 × 105 cells/mL in a four-well dish pre-coated with 0.5% gelatin (MilliporeSigma, Burlington, MA, USA) in culture medium before their utilization for pEPSC seeding.
Derivation and culture of pEPSCs
To remove the zona pellucida, porcine IVF, PA, and SCNT blastocysts were incubated in a 0.5% protease solution for 1 min. For plating, intact day-6 or -7 blastocysts were washed and plated directly onto feeder layers under a microscope (SMZ645; Nikon, Tokyo, Japan) and cultured in modified pEPSCs medium (pEPSCM). The modified pEPSCM was prepared by incorporation using basal medium in a 1:1 mixture of mTeSRTM1 (100–0276; STEMCELL Technologies, Vancouver, BC, Canada) and DMEM/F-12 (Gibco) supplemented with 1× N2 supplement (Thermo Fisher Scientific), 1× B27 supplement (Thermo Fisher Scientific), 1× NEAA, 1× GlutaMAX supplement (Gibco), 1 × β-mercaptoethanol, 0.5 µM CHIR99021 (S1263; Selleck Chemicals, Houston, TX, USA), 0.3 µM WH-4-023 (S7565; Selleck Chemicals), 2.5 µM XAV939 (X3004), 65.0 µg/mL vitamin C (50-81-7), 10.0 ng/mL leukemia inhibitory factor (LIF) (250-02; PeproTech, Cranbury, NJ, USA), 20.0 ng/mL Activin A (78001; STEMCELL Technologies), 0.3% FBS, and 0.5× Anti-anti (Gibco). After 48 h, the attachment efficiency of primary cultures was determined by scoring the number of attached colonies. After 5–7 days of culture, the primary outgrowth of EPSC-like cells was mechanically dissociated into several clumps using pulled-glass pipettes under a stereomicroscope (Nikon). The clumps were then re-seeded on fresh feeder cells, and subsequent EPSC lines were passaged mechanically every 3–4 days using TrypLE Express (12605010; Gibco) after washing briefly in Hanks’ balanced salt solution (14170-112; Gibco). The medium was replaced daily, and all cells were cultured at 37 °C with 5% CO2 under humidified conditions.
Cultivation of porcine induced pluripotent stem cells (iPSCs)
The porcine iPSCs used in this study were kindly supplied by Professor Jongpil Kim from Dongguk University, Republic of Korea. These cells were generated by transfecting porcine embryonic fibroblasts with five doxycycline-inducible human factors: FUW-tetO-hOct4, FUW-tetO-hSox2, FUW-tetO-hKlf4, FUW-tetO-hc-Myc, and FUW-M2rtTA, all acquired from Addgene (Watertown, MA, USA) and delivered via lentiviral vectors. iPSCs were cultured at 37 °C in an atmosphere of 5% O2 and 95% air on feeder layers using stem cell basal medium [a 1:1 mixture of DMEM (Gibco) and F10 (Gibco), supplemented with 15% FBS, 1× GlutaMAX, 1× ß-mercaptoethanol, 1× MEM-NEAA, and 1× Anti-anti] containing 10.0 ng/mL LIF (250-02; PeproTech) and 2 µg/mL doxycycline (D9891). The culture medium was refreshed daily, and the cells were passaged every 3 days. Passaging was performed using a 0.04% trypsin treatment, which allowed the cells to maintain a stable morphology for over 50 passages.
Derivation and culture of porcine NT-derived embryonic stem cells (ESCs) using cloned embryos
To plate the cloned blastocysts, the procedure outlined above for deriving pEPSCs was performed. Following a culture period of 10–14 days, ES-like primary colonies were obtained and subsequently mechanically dissociated into multiple clumps using pulled-glass pipettes. These clumps were then re-seeded onto a fresh feeder layer, and the resulting ES cell lines were passaged mechanically at 5–9 days intervals without enzymatic treatment. Upon reaching a size of 3–4 mm, the colony was carefully detached from the feeder layer and transferred into a drop of ESC culture medium comprising stem cell basal medium, as mentioned above, supplemented with 4 ng/mL basic fibroblast growth factor (Invitrogen, Carlsbad, CA, USA), where it was dissociated into smaller clumps. The clumps were subsequently placed in a new feeder layer supplemented with fresh ESC culture medium. The medium was refreshed daily, and the cells were cultured in a humidified atmosphere maintained with 5% CO2 at 37 °C.
Gene expression analysis by qPCR
For gene expression analysis, stem cells or embryoid bodies (EBs) were washed twice in dPBS (Gibco) and stored at − 80 °C until analysis. Total RNA was extracted using RNAiso Plus reagent (TaKaRa Bio Inc., Otsu, Japan), followed by complementary DNA (cDNA) synthesis using Reverse Transcription 5× Master Mix (Elpis Bio, Inc., Daejeon, Republic of Korea) in accordance with the manufacturer’s instructions. Next, 1 µL of the synthesized cDNA was mixed with 10 µL 2× SYBR Premix Ex Taq (TaKaRa Bio Inc.) and 10 pmol of specific primers (Macrogen, Daejeon) (Additional file 1) and subjected to qPCR using a CFX96 Touch Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, CA, USA). Reactions were performed as follows: 40 cycles of denaturation at 95 °C for 15 s, annealing at 57 °C for 15 s, and extension at 72 °C for 15 s. Relative quantification was performed using threshold cycle (Ct)-based methodologies, maintaining a constant fluorescence intensity. The relative mRNA expression (R) was calculated using the equation R = 2−[∆Ct sample − ∆Ct control]. The R-values obtained for each gene were quantified relative to those of GAPDH.
Immunofluorescence analysis
After washing twice with dPBS containing calcium and magnesium ions (LB 001–01; Welgene Biotech, Taipei City, Taiwan), cells grown in 8-well chamber slides (154534; Thermo Fisher Scientific) were fixed with 4% paraformaldehyde (PFA) for 10 min. Subsequently, the cells were incubated with blocking buffer (12411 S; Cell Signaling Technology, Danvers, MA, USA) for 1 h at room temperature (RT) and labeled with primary antibodies (shown in Additional file 2) overnight at 4 °C. Afterward, the cells were washed thrice with dPBS containing 0.2% Tween-20 and incubated with the appropriate secondary antibodies for 1 h at RT. After three washes, nuclei were stained with Hoechst 33342 and mounted using Vectashield (Vector Laboratories, Burlingame, CA, USA). Stained cells were imaged using a confocal microscope (Carl Zeiss, Oberkochen, Germany) and ZEN 2009 Light Edition software (Carl Zeiss).
Alkaline phosphatase (AP) staining
For AP activity detection, pEPSCs were fixed in 4% PFA for 5–10 min. After washing with 0.1 M Tris-HCl (pH 9.5) solution twice, they were stained with a solution containing nitro blue tetrazolium chloride/5-bromo-4-chloro-3-indolyl phosphate toluidine salt stock solution (Roche) for 30 min. The stained cells were analyzed under a microscope (Nikon). Staining was performed at RT.
Karyotyping
To induce metaphase, confluent monolayer pEPSCs were exposed to 10 µg/mL colcemid (Gibco) for 3–5 h. Subsequently, karyotyping analysis was performed as described previously [32]. Finally, the chromosomes on the slides were counted and observed under a light microscope (Nikon) to assess cytogenetic abnormalities.
BLS and EB formation via spontaneous differentiation
Colonies of the pEPSCsNT line were dissociated into single cells using TrypLE Express Reagent (Gibco) and subsequently cultured in suspension on a 35 mm low-attachment plate containing the aforementioned cell culture medium to promote spontaneous differentiation. For the initial 24 h of culture, the medium was supplemented with 10 µM ROCK inhibitor (Y-27632; ROCKi); thereafter, the medium was replaced with ROCKi-free medium, with subsequent medium changes every 2 days. After a culture period of 7 days, the blastocyst-like structures (BLSs) were fixed directly using 4% (v/v) PFA solution for 10 min, following a wash with dPBS for subsequent immunofluorescence staining. Concurrently, a subset of EBs was collected and plated onto eight-well chamber slides (Thermo Fisher Scientific) coated with 0.1% (v/v) gelatin for adherent culture in cell culture medium. After an additional 10 days of culture, the adherent cells were fixed with 4% (v/v) PFA for subsequent immunofluorescence staining. The remaining EBs were cultured for an additional period of up to 46 days before being fixed for immunohistochemical staining.
Cryosectioning and immunohistochemistry using day-46 EBs
The fixed EBs were washed at least thrice with dPBS and subsequently immersed in a 30% sucrose–PBS solution within a microcentrifuge tube for incubation overnight at 4 °C. Following an additional 2–3 washes, the EBs were placed in a microcentrifuge tube with dPBS and dispatched to OBEN Bio Inc. (Suwon, Republic of Korea) for cryosectioning. The slide-affixed EB sections were stained according to the procedure outlined in the section titled “Immunofluorescence analysis.” During staining, the section slides were circumscribed with a PAP Pen to create a hydrophobic barrier (ImmEdge™; Vector Laboratories) and placed in a humid chamber. All subsequent procedures were performed at RT in a humid environment.
Transcriptome analysis
pEPSCsNT, iPSCs, and cloned blastocysts that developed to day 7 were washed twice with dPBS, pelleted by centrifugation, and immediately frozen in liquid nitrogen. The samples were then sent to Theragen Etex, Inc. (Seongnam, Republic of Korea) for RNA extraction, cDNA library preparation, and whole-transcriptome sequencing using an Illumina NovaSeq6000 platform (Illumina, San Diego, CA, USA). For porcine data, Ensembl build Sscrofa11.1 (GCA_000003025.6) was used. Libraries were constructed for 151 bp paired-end sequencing using a SMARTer Stranded Total RNA-Seq Kit V2-Pico Input (Takara Bio, Inc.). Before generating single-stranded cDNA, rRNA was removed. The integrity of the total RNA was evaluated using an Agilent 2100 BioAnalyzer (Agilent Technologies, Santa Clara, CA, USA; Additional file 3), and cDNA libraries were quantified using a KAPA library quantification kit (Kapa Biosystems, Wilmington, MA, USA).
RNA-seq data of porcine in vivo and cloned embryos at various developmental stages, as published by He et al. [24], were downloaded from the Gene Expression Omnibus (GEO) database (Accession no. GSE125706). The data were used to perform principal component analysis and Pearson’s correlation coefficient analysis, facilitating a comparative assessment of our sample data. The expression of the specific genes of interest was extracted from the expression matrix and visualized as a heatmap using GraphPad Prism 10.1 (GraphPad Software, La Jolla, CA, USA). After assessing the differentially expressed genes (DEGs), volcano plots were generated by Theragen Etex, Inc., and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were evaluated using the DAVID database (https://david.ncifcrf.gov).
Plate coating with various ECMs
In this study, we tested several ECM products, including Matrigel (354277; Corning Life Sciences, Corning, NY, USA), fibronectin (33010-018; Thermo Fisher Scientific), Laminin-521 (A29248; Gibco), and vitronectin (VTN)-N (A14700; Gibco), to optimize feeder-free culture conditions for pEPSCsNT line. The coating process was performed according to each manufacturer’s instructions. After plating the extracellular matrix (ECM)-coated plates, pEPSCsNT were dissociated using TrypLE Express (Gibco) and assessed at 1.5, 24, and 72 h to analyze the adherence rate and survival index by cell counting.
Differentiation of pEPSCsNT cultured under feeder-free conditions to trophoblast lineages
After dissociation with TrypLE Express (Gibco), pEPSCsNT cultured under feeder-free conditions (FF-pEPSCsNT) were plated onto a Matrigel-coated plate with DMEM/F12 (Gibco) supplemented with 20% (v/v) knockout serum replacement (10828028; Gibco) and 10 µM ROCKi for a day. On the second day, the medium was replaced by 20% (v/v) knockout serum replacement basal media without ROCKi containing 10 µM SB431542 (S4317), 50 ng/mL BMP4, and 0.1 µM PD032590 (FGF receptor inhibitor) to induce trophoblast differentiation. On day six of differentiation, the cells were collected for analysis.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 10.1 (GraphPad Software). The results are presented as means ± standard error of the mean. Each experiment was repeated at least thrice unless a different number of replicates is stated in the legend. Statistical evaluations were performed using an unpaired two-tailed Student’s t-test or ANOVA, as detailed in the figure legends. P < 0.05 was considered statistically significant. The statistical methods, P-values, and sample sizes are provided in the figure legends.
The work has been reported in line with the ARRIVE guidelines 2.0.