

Large-scale genetic characterization nominates known and novel potential disease-causing variants associated with Alzheimer’s disease and related dementias
We used three different datasets (AoU, UKB, and 100KGP) to identify known and novel potential disease-causing variants. A summary of the variants that were identified can be found in Fig. 3 and Supplementary Fig. 1. Within the AoU dataset (discovery phase), we identified a total of 193 variants in 11 genes (APP, PSEN1, PSEN2, TREM2, GRN, MAPT, GBA1, SNCA, TBK1, TARDBP, and APOE). All variants and their allele frequencies in cases and controls across different ancestries are available in Supplementary Data 1. Among these variants, 36 were present only in cases and had a Combined Annotation Dependent Depletion (CADD) score > 20 (CADD score > 20 means that the variant is among the top 1% most pathogenic in the genome, as a proxy for its deleteriousness). All 36 identified variants were heterozygous. Of these, five were previously reported in AD or FTD (Table 1), while 31 were novel (Table 2). Of the five known variants, three were found in cases of European ancestry, one in cases of African ancestry, and one in cases of American Admixed ancestry. Among the 31 novel variants, 20 were found in cases of European ancestry, four in cases of African ancestry, two in cases of American ancestry, two in a case of Ashkenazi Jewish ancestry, and three in cases of African Admixed ancestry.
Fig. 3: Summary of known and novel variants identified during the discovery phase.
Variants in red are known, while variants in black are novel. † Singleton variants identified in the discovery phase; ‡ Variants found in more than one individual during the discovery phase; * Variants replicated across biobanks in the discovery phase; # Variants replicated only in cases during the replication phase across biobanks.
Table 1 Discovery phase: Multi-ancestry summary of known potential disease-causing variants only present in Alzheimer’s disease and related dementia cases in AoU, 100KGP and UKBTable 2 Discovery phase: Multi-ancestry summary of novel potential disease-causing variants only present in Alzheimer’s disease and related dementia cases in AoU, 100KGP and UKB
Within the UKB (discovery phase), we identified a total of 815 variants in the APP, PSEN1, PSEN2, TREM2, GRN, MAPT, GBA1, SNCA, TBK1, TARDBP, and APOE genes (Supplementary Data 2). Among these, 121 variants were present only in cases and had a CADD score > 20. All 121 identified variants were heterozygous. Of these, 20 were previously reported as disease-causing in AD, FTD, frontotemporal lobar degeneration (FTLD), and Gaucher disease (Table 1), while 101 were novel (Table 2). A majority (n = 113) of the variants were identified in individuals of European genetic ancestry, while three were identified in cases of African ancestry, three in cases of South Asian ancestry, and one each in cases of East Asian and Ashkenazi Jewish ancestries. The allele frequencies of the variants across different ancestries are reported in Supplementary Data 2.
Within the 100KGP data (discovery phase), we identified a total of 13 variants in the APP, PSEN1, PSEN2, GRN, GBA1, TBK1, and TARDBP genes (Supplementary Data 3). Among cases, no variants were identified in the MAPT, TREM2, SNCA, and APOE genes. Of the 13 variants, four were only present in cases and had a CADD score >20. All four identified variants were heterozygous and previously reported in individuals of European ancestry. Among these four variants, PSEN1 p.R269H and TARDBP p.G287S had been previously reported as causes of AD and amyotrophic lateral sclerosis (ALS), respectively (Table 1), while the remaining two variants in the APP and PSEN2 genes were novel (Table 2). The allele frequency of each variant is presented in Supplementary Data 3.
We next used the ADSP and AMP PD cohorts to replicate the disease-associated variant findings listed above.
Replication analyses support the relevance of identified genetic variation across diverse ancestries
Nine variants identified in AoU, 20 identified in UKB, and four identified in 100KGP were replicated in AD cases in the ADSP cohort (Supplementary Data 4). Below we detail these variants and their occurrences in both European and non-European participants.
Among the nine variants found in AoU that were replicated in ADSP, three variants—APP p.A713T, PSEN1 p.A79V, and TARDBP p.G287S — had been previously reported, while six variants—APP p.L597W, MAPT p.G701R, MAPT p.G750S, SNCA p.Q99R, TBK1 p.N42S, and TBK1 p.R507I—were novel. The APP p.L597W variant was found in African, African Admixed, and Complex Admixture History ancestries. Searching for other dementia cases resulted in the identification of APP p.A713T in one DLB case and PSEN1 p.A79V in a possible AD case according to ADSP diagnosis criteria. The allele frequency of each variant per genetic ancestry in cases and controls is reported in Supplementary Data 4.
Among the four variants identified in the 100KGP dataset that were replicated in the ADSP cohort, PSEN1 p.R269H and TARDBP p.G287S were previously reported, while PSEN2 p.D320N and APP p.Y407H were novel. We observed the previously reported PSEN1 p.R269H variant in five cases and no control participants. This variant was found in European ancestry individuals from the 100KGP cohort and was also observed in individuals of African Admixed ancestry (two cases) and European ancestry (three cases) in the ADSP dataset. TARDBP p.G287S was found in one European control individual and absent in cases. Of the novel variants, the PSEN2 p.D320N variant was found in five controls and was not observed in any cases, while the APP p.Y407H variant was observed in two cases and one control. Searching for additional cases led to the discovery of PSEN1 p.R269H in a patient with mild cognitive impairment (MCI).
Among the 20 variants identified in the UKB cohort that were replicated in the ADSP dataset, six variants—PSEN1 p.A79V (five cases and one control), PSEN1 p.R269H (five cases), GRN p.R493X (three cases), MAPT p.R406W (four cases and one control), GBA1 p.W223R (one case and one control), and TBK1 p.R228H (one case)—have been previously reported. The remaining 14 variants were novel. Most of the novel variants were found in European cases in the UKB. Among the known variants, the PSEN1 p.R269H variant was found in cases of both African Admixed and European ancestries, and GBA1 p.W223R was found in a case of Complex Admixture History ancestry and a control of African ancestry. The four remaining known variants were observed in individuals of European ancestry in the ADSP cohort.
Novel variants identified in non-European participants include PSEN1 p.R220Q (one African case, two European cases, and one African Admixed control), PSEN1 p.T291A (one African Admixed case, one American Admixed control, and two controls with Complex Admixture History), MAPT p.A60G (two African Admixed cases, one African Admixed control, two African controls, and four controls with Complex Admixture History), GRN p.V490M (one African Admixed case), MAPT p.R103W (one American Admixed control), APP p.P251S (one South Asian control), APOE p.Q65X (one African control), and APOE p.E275G (one African Admixed control). Searching for other cases resulted in the identification of PSEN1 p.A79V in one possible AD patient, PSEN1 p.R269H and MAPT p.A60G in two independent MCI patients, and GBA1 p.W223R and APP p.A209T in two independent progressive supranuclear palsy (PSP) patients (Supplementary Data 4).
We identified a novel SNCA variant (p.Q99R) in the AoU dataset, while the UKB dataset revealed two additional variants in SNCA: p.P90H and p.A91S. Both p.P90H and p.A91S were predicted to be likely pathogenic according to prediction estimates and have not been previously reported as disease-causing. Notably, the SNCA p.Q99R variant was replicated in the ADSP cohort. All three variants were heterozygous, and none of these variants were found in any controls across these datasets. However, the age at onset (AAO) of these variant carriers is not consistent with a potential disease-causing highly deleterious effect.
Our analyses of multiple datasets identified 12 variants (three from AoU, eight from UKB, and one from 100KGP) that were absent in the ADSP control cohort.
Across each of our discovery datasets, we identified six candidate variants—APP p.A713T, MAPT p.G750S, GRN p.V490M, GRN p.R493X, APP p.D516N, and TARDBP p.G287S—present in AMP PD (DLB cases and controls). GRN p.V490M and TARDBP p.G287S were present in one control and absent in cases, GRN p.R493X was present in one case and absent in controls, while the other three were present across both cases and controls. The allele frequencies of these variants are detailed in Supplementary Data 5.
Among the 156 variants identified in this study, 18 were found exclusively in non-European ancestries. Notably, APPp.L597W and MAPTp.A60G were replicated in African and African Admixed ancestries across different datasets. These data highlight the potential significance of these variants in groups that are often underrepresented in genomic studies.
Supplementary Fig. 2 shows the allele frequencies of all identified known and novel variants with a CADD > 20 in the discovery and replication phases across all ancestries in each biobank.
Previously reported disease-causing variants raise questions about potential pathogenicity
In addition to identifying potential disease-causing variants, we also identified variants in SNCA, APP, TARDBP, GRN, and GBA1 that may not cause disease or may be risk factors of incomplete penetrance based on their presence in control participants.
Although the SNCA p.H50Q variant was initially identified as a disease-causing mutation in Parkinson’s disease (PD)16, subsequent research has challenged its pathogenicity17. Our study confirms that it is not pathogenic across other synucleinopathies such as DLB, based on its occurrence in five European controls in AoU and 28 European controls in UKB (Supplementary Data 6).
Additionally, several research studies have reported the APP p.A713T variant to be disease-causing18,19. In our study, we found this variant in heterozygous state in five control individuals: two in UKB, two in ADSP, and one in AMP PD. Interestingly, the APP p.E665D variant, which has been widely reported to cause AD20,21, was found in one control in AoU in her late 70s. However, it is possible that the variant shows incomplete penetrance, that this individual may develop disease later in life, or that she may even harbor unidentified resilient genetic variation. Another previous study evaluating the role of APP p.E665D also questioned the pathogenicity of this variant22.
TARDBP p.G287S, previously reported as disease-causing for ALS23, was found in five controls—three in UKB, one in ADSP, and one in AMP PD. Similarly, TARDBP p.N390S, also previously reported as disease-causing for ALS24, was found in two controls in UKB.
GBA1 coding variants generally exhibit incomplete penetrance and act as genetic risk factors in heterozygous state. Homozygous GBA1 variants, including the p.T75del and c.115+1G>A mutations, have been reported to cause Gaucher disease25,26,27,28. We found these two variants in heterozygous state in one case and one control in AoU. GBA1 p.T75del was found in individuals of African ancestry, and GBA1 c.115+1G>A was found in individuals of European ancestry in both a case and a control. The GBA1 c.115+1G>A variant was also found in nine European controls in the UKB cohort. We identified 13 additional heterozygous variants in GBA1 (p.R502C, p.A495P, p.L483R, p.D448H, p.E427X, p.G416S, p.N409S, p.R398X, p.R296Q, p.G241R, p.N227S, p.S212X, and p.R159W) that have been reported as disease-causing for Gaucher disease in homozygous state. Three variants in GRN, including two loss of function variants (p.Q130fs and p.Y294X) and one splicing variant (c.708+6_708+9del), have been previously reported to cause FTD, FTLD, and neurodegenerative disease25,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43. Each of these 16 variants were found in several control individuals. The frequency and number of carriers for each variant in cases and controls are reported in Supplementary Data 6.
Genetic-phenotypic correlations provide valuable clinical insights
Clinical data for the identified variants are summarized in Supplementary Data 7 and 8. Here, we briefly explain the main findings.
The GRN p.R493X variant is the most reported pathogenic mutation in this gene. This variant has been associated with several types of dementia, including FTD, FTLD, primary progressive aphasia, AD, and corticobasal degeneration. It is known to be more frequently identified among FTD cases, particularly in early-onset forms44. In one study investigating the genetics underlying disease etiology in 1118 DLB patients, this variant was reported in a single case, presenting a wide range of neurological phenotypes that could not lead to a conclusive diagnosis. Severe dementia, parkinsonism, and visual hallucinations suggested a clinical diagnosis of AD or mixed vascular dementia. However, the final neuropathological diagnosis was suggested to be AD, DLB, and argyrophilic grain disease45. We identified this variant in four European AD patients, three of whom presented with early onset in their fifties. Interestingly, we also identified this variant in a DLB patient in her early 60s. Neuropathological data and McKeith criteria46 strongly supported a diagnosis of DLB in this patient. Although this variant has been widely reported across different types of dementia, our finding is the first report of this variant in DLB with a McKeith criteria of “high likelihood of DLB,” expanding the etiological spectrum of GRN variation (Supplementary Data 7).
GRN p.C222Y was previously reported in a familial AD case from Latin American (Caribbean Hispanic) ancestry47,48. While the AAO for this patient was not reported, the mean AAO for the cohorts under study was 56.9 years (SD = 7.29), with a range of 40–73 years. In our study, we identified this variant in an individual of American Admixed ancestry with dementia in his late 40s and a disease duration of 11 years to date. This finding reinforces the role of this variant in early-onset disease.
There are several other interesting findings regarding variants in GRN. The GRN c.708+1G>A variant was previously reported in several FTD, FTLD, and corticobasal syndrome (CBS) cases, mostly early-onset43,49. We identified this variant in two European AD cases, both diagnosed in their 70s, marking the first report of this variant in late-onset Alzheimer’s disease (LOAD). The GRN p.P166fsX variant was previously reported in an early-onset behavioral variant FTD case50. In our study, we identified this variant in a European dementia case diagnosed in her mid 70s with a disease duration of eight years to date. Furthermore, the GRN p.R418X variant is identified in the literature in two cases of FTLD with ubiquitin-positive inclusions (FTLD-U) with AAOs of 49 and 60 years51. We identified this variant in a European dementia case in her early 70s. Both findings represent the first report of these variants in late-onset dementia.
PSEN1 R269H is a known pathogenic variant causing early-onset Alzheimer’s disease (EOAD)52,53. However, it has been previously reported in only two LOAD cases54,55. In our study, we identified this variant in European and African Admixed ancestries in a total of 12 cases (eight AD and four related dementias), six of which were early-onset (≤65 years) and six were late-onset (>65 years). This finding underscores the potential for PSEN1 p.R269H to contribute to LOAD with reduced penetrance. Additionally, one EOAD case that presented with hallucinations56 and another case that manifested a behavioral presentation57 have been reported to carry this variant. In this study, we identified PSEN1 p.R269H in one FTD patient in the 100KGP cohort, marking the first report of this variant in FTD.
MAPT p.R406W has been reported in several familial cases of FTD with parkinsonism, all with early onset58. There are only two articles related to this variant in AD. The first describes a family with AD-like symptoms, with an average AAO of 61 years59, and the other reports a familial AD case with an AAO of 50 years60. In our study, we identified this variant in nine AD cases, with a mean AAO of 61 years. This finding underscores the role of this variant in EOAD.
TARDBP p.G287S has been reported as a cause of ALS23. Here, for the first time, we identified this variant in two early-onset dementia cases in the AoU and 100KGP datasets.
TBK1 p.D639H was previously reported in EOAD61. Here, we identified this variant in a LOAD patient in her late 70s. TBK1 p.N22H, p.R228H, and p.E476fs were previously reported in ALS, ALS, and motor neuron disease, respectively62,63. We identified these variants in AD cases for the first time.
Several variants in GBA1, such as p.F298L, p.V230G, p.W223R, and p.P198L, have been previously reported in Gaucher disease patients. In our study, we identified these variants in heterozygous state in one AD case and five dementia cases, all with late onset. GBA1 p.W223R was found in one AD case of Complex Admixture History ancestry. GBA1 mutations are known to confer an increased risk for dementia in PD and DLB. Notably, they have not been previously suggested to contribute to AD.
Similarly, APP p.E693Q has been reported in a few AD cases. In our study, we identified it in a related dementia case and no controls. This finding suggests that this variant may also be implicated in other types of dementia.
Several known variants identified in this study confirm previous findings related to disease type and onset. For example, the APP p.V717L variant has been reported in numerous AD cases, primarily in early-onset forms64,65. In our study, we identified this variant in two cases of EOAD with AAO ranges of 51–55 years and 56–60 years, respectively. Additional examples are reported in Supplementary Data 7 and 8.
In AoU, the SNCA p.Q99R variant was found in a female patient diagnosed in her late 60s with unspecified dementia without behavioral disturbance. In ADSP, the variant was identified in a male patient with pure AD in his early 70s. SNCA p.P90H and p.A91S were found in two males in their late 70s in the UKB cohort. All four patients were of European ancestry. Previously reported mutations in SNCA are known to cause early-onset PD and DLB66,67. The mean AAO in patients carrying SNCA mutations in this study is 72.75 years. These data suggest that these variants may not be disease-causing but could represent rare risk factors given their absence in controls and the replication of p.Q99R across datasets.
Novel variants found in this study that may potentially be associated with early-onset dementia include: p.L597W, p.V375I, p.L364F, p.A209T, p.D460N, p.R409C, and p.V227L variants in APP; p.R54X and p.M457V in PSEN1; p.H169R, p.D320N, and p.G349R in PSEN2; p.R556C and p.V28fs in GRN; p.G332LfsX64 and p.G701R in MAPT; p.G103D and p.A42T in GBA1; p.R556C in TARDBP, p.N42S and p.R507I in TBK1; p.E275G and p.D271N in APOE and TREM2 p.W44X. Among these variants, APP p.L364F and PSEN1 p.R54X were found in vascular dementia cases with AAO ranges of 56–60 years and 41–45 years, respectively. Additionally, PSEN2 p.D320N was found in an FTD case with an AAO of mid-50s. Another notable finding is the identification of GRN p.R556C in a dementia case with an AAO of mid-30s.
APOE drives different population-attributable risk for Alzheimer’s disease and related dementias
We separately analyzed ancestry-specific effects of APOE on AD/ADRDs. The summary of these findings is depicted in Fig. 4, Supplementary Data 9, and Supplementary Fig. 3.
Fig. 4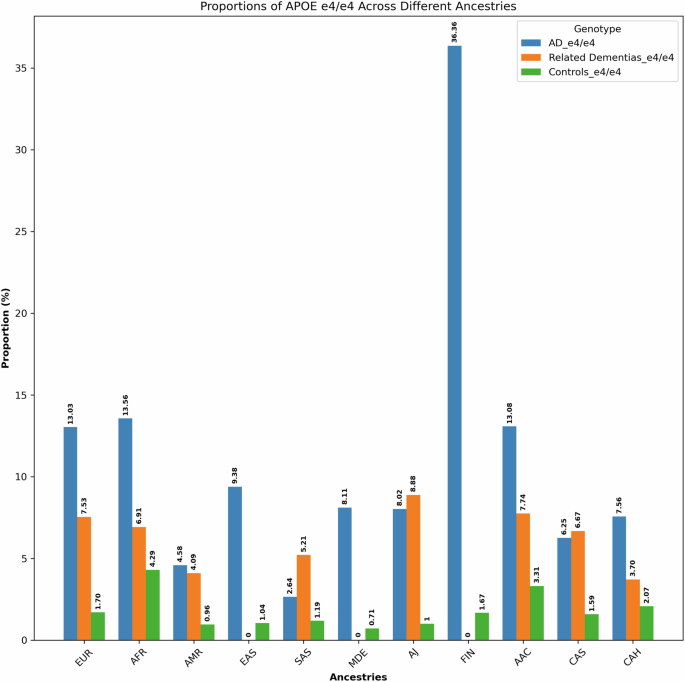
Proportions of APOE ε4/ε4 across 11 genetic ancestries in Alzheimer’s disease, related dementias, and controls in all datasets.
In AoU, UKB, and 100KGP datasets, the APOE ε4/ε4 genotype exhibits a higher frequency among both AD patients and control individuals of African and African Admixed ancestries compared to Europeans. Related dementia patients show similar results in UKB. In AoU, related dementia cases of African Admixed ancestry show a higher frequency than cases of European ancestry, while case frequencies are similar between Africans and Europeans, likely due to the limited number of individuals with African ancestry in this dataset. In ADSP, the frequencies of this genotype among AD patients are similar across the three ancestries. Among control individuals in ADSP, APOE ε4/ε4 is more frequent in African Admixed and African ancestries than in Europeans, as previously reported68. Notably, the APOE ε4/ε4 genotype was absent from African and African Admixed DLB cases and controls in the AMP PD dataset. The frequency of APOE ε4/ε4 in Europeans was higher in cases compared to controls in AMP PD.
When combining results across all datasets, the frequency of APOE ε4/ε4 in African and African Admixed AD patients was still higher than in Europeans, but the values were not significantly different. However, the frequency of APOE ε4/ε4 in control individuals of African and African Admixed ancestries was substantially higher than in controls of European ancestry. Additionally, the frequency of APOE ε4/ε4 in Finnish individuals was higher in AD cases and lower in controls compared to Europeans.
Disease-modifying variants in APOE ε4 carriers modulate Alzheimer’s and dementia risk across different ancestries
The summary of our findings for the frequencies of protective and disease-modifying variants under study, alongside APOE genotypes across all five datasets, is depicted in Supplementary Data 10–14. The proportions of individuals carrying APOE ε4 homozygous or heterozygous genotypes alongside protective or disease-modifying variants, within the total population, total ε4/ε4 carriers, and total ε4 carriers across each ancestry, combined across all biobanks, in AD, related dementias, and controls are reported in Supplementary Fig. 4 and Supplementary Data 15. Summaries of our findings for all the assessed models in APOE ε4, ε4ε4, and ε3ε3 are shown in Supplementary Fig. 5, and Supplementary Data 16–25.
In brief, we observe higher frequencies of individuals carrying APOE:rs449647-T, 19q13.31:rs10423769-A, APP:rs466433-G, or APP:rs364048-C protective variant alleles alongside either one or two copies of APOE ε4 among African and African Admixed ancestries compared to Europeans for AD, related dementias, and controls. Carriers of APOE:rs449647-T and 19q13.31:rs10423769-A are particularly noteworthy because APOE:rs449647-T displays the highest frequency among these ancestries, and the ratio of frequencies for 19q13.31:rs10423769-A in both African and African Admixed ancestries compared to Europeans is substantially higher than the other protective/disease-modifying variants investigated across all three cohorts. In individuals of African ancestry, 19q13.31:rs10423769-A was found to have a higher frequency in controls compared to both AD and related dementia cases among APOE ε4 homozygous or heterozygous carriers. In contrast, APOE:rs449647-T was found to have a lower frequency in controls compared to AD cases among APOE ε4 homozygous or heterozygous carriers in African ancestry but showed a higher frequency in controls carrying APOE ε4/ε4 versus cases in European populations (Supplementary Data 15).
The combination of TOMM40:rs11556505-T with either homozygous or heterozygous APOE ε4 was observed to have a higher frequency in Europeans and a lower frequency in Africans compared to most other ancestries across all three phenotypes. Additionally, the combination of NOCT:rs13116075-G and APOE ε4 homozygous or heterozygous was found to have a higher frequency in individuals of European and African Admixed ancestry versus Africans, in AD cases as compared to controls.
The protective model shows a modifying effect of APOE:rs449647-T in European, African, and Ashkenazi Jewish ancestries as well as a modifying effect of TOMM40:rs11556505-T in European, American Admixed, Ashkenazi Jewish, African Admixed, and individuals of Complex Admixture History. The R² model indicates that these variants are not in linkage disequilibrium with the APOE risk variants rs429358 and rs7412.
Significant interactions were found between APOE ε4 and the following variants: 19q13.31:rs10423769-A in Africans; NOCT:rs13116075-G in both African and African Admixed populations; CASS4:rs6024870-A in the Complex Admixed History group; LRRC37A:rs2732703-G in the American Admixed ancestry; APOE:rs449647-T in European and African Admixed ancestries; and TOMM40:rs11556505-T in Europeans. In APOE ε4/ε4 carriers, the interaction with APOE:rs449647-T was found to be significant in American Admixed and African Admixed populations, while TOMM40:rs11556505-T was significant in the European ancestry.
The interaction model in APOE ε3/ε3 shows no significant p value for 19q13.31:rs10423769-A in Africans, NOCT:rs13116075-G in African Admixed ancestry, CASS4:rs6024870-A in Complex Admixed History, and LRRC37A:rs2732703-G in American Admixed ancestry, but highly significant p values for APOE:rs449647-T and TOMM40:rs11556505-T in European and NOCT:rs13116075-G in Africans, with opposite directional effects compared to APOE ε4 carriers. These data confirm the role of these variants in modulating the effect of APOE ε4 in AD risk.
To assess the potential enrichment of protective or disease-modifying variants in cases versus controls, we conducted logistic regression on individuals with the highest genetic risk burden based on the PRS, including 1,997 cases and 559 controls. Results are presented in Supplementary Data 26. The analysis revealed no significant differences between cases and controls for these variants and indicated no interaction between these variants and PRS to modify disease penetrance. This analysis may be underpowered due to the low frequency of these variants.
We performed a burden analysis to determine whether there was an enrichment of predicted pathogenic variants in cases compared to controls by gene, after excluding known variants. Burden testing, adjusted for sex, age, and principal components (PCs 1–10) using SKAT-O, revealed an enrichment of rare variants in GBA1 (P = 0.023) (Supplementary Data 27 and 28).