
Animal care
Animal care, handling, and experimental procedures were conducted in compliance with protocols approved by local government authorities (Bezirksregierung Köln). Permission to maintain and breed mice was issued by the Department for Environment and Consumer Protection, Veterinary Section, Cologne, North Rhine-Westphalia, Germany. All mice were housed in individually ventilated cages at 22–24 °C at constant humidity (50–70%) with a 12-h light–dark cycle with ad libitum food and water unless otherwise stated for experimental conditions.
Mouse models
C57BL/6NCrl mice were purchased from Charles River. AgRP-IRES-Cre mice (JAX stock, #012899)65, M71-IRES-Cre (JAX stock, #006677)83, OMP-Cre (JAX stock, #006668)83 and R26-LSL-ReaChR-mCitrine (JAX stock, #026294)54 were originally obtained from The Jackson Laboratory. Cre lines were maintained heterozygous through breeding to wild-type C57BL/6NCrl (Charles River), while R26-LSL-ReaChR-mCitrine mice were maintained as homozygous stock breeding at the Max Planck Institute for Metabolism Research.
Diets
All diets used were obtained from ssniff Spezialdiäten (ssniff). NCD (ssniff R/M-H-Phytoestrogenarm) was used as the control maintenance diet and contained 57% calories from carbohydrates, 34% from protein and 9% from fat. The BFD, the PB-FD and AFD are isonutritional diets to NCD, containing 0.15% artificial flavours (Elli’s Aromen; bacon, AS10301; peanut butter, AS10312) or 1% acetophenone (AFD, used in behavioural tests). HFDs used in this study are: HFDlard (ssniff EF acc. D12492 (I) mod, which contains 21% calories from carbohydrates, 19% from protein and 60% from fat derived from pork lard); HFDbutter (ssniff EF acc. D12492 (I) mod, which contains 21% calories from carbohydrates, 19% from protein and 60% from fat derived from butter) and HFDacetophenone containing 0.25% acetophenone in HFD (HFDlard; ssniff EF acc. D12492 (I) mod). The peanut butter-based HFD (HFDpeanut-butter) containing 34.9% kcal from fat was produced in-house by mixing 40% organic peanut butter (Bio Erdnussmus, dm-Drogerie Markt) with 60% NCD (ssniff R/M-H-Phytoestrogenarm) previously ground using a Koenic food blender. Feeding preference tests were performed using control diet (CD; ssniff EF acc. E15748) containing 67% calories from carbohydrates, 20% of protein, and 13% of fat.
Paradigm of developmental exposure to fat-related sensory cues through the diet
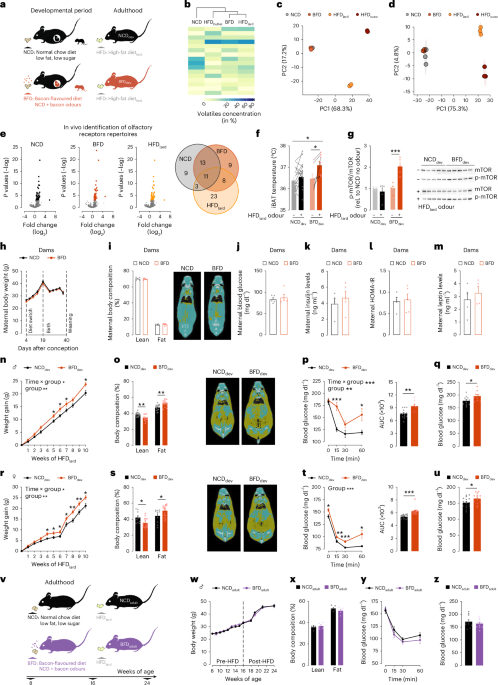
C57BL/6NCrl mice were ordered from Charles River between 5 and 6 weeks old and allowed to acclimate in a group setting of 4–5 mice per cage for 2–3 weeks. To minimize the increased risks of pup mortality reported in primiparous C57BL/6N mice, experimental cohorts of animals were generated using pregnancy-experienced females that have already undergone one pregnancy at 8 weeks old. Mice were paired one male to one female for breeding (1:1 breeding) to generate experimental cohorts. Male breeders were C57BL/6NCrl or AgRP-IRES-Cre heterozygotes for photometry experiments. Males were removed upon first signs of visible pregnancy and single caged. Females taking longer than 19 days to show visible signs of pregnancy were excluded. At postnatal day 1 (P1; day of birth considered as P0) pups were removed from the cage and tissue collected. One week later, males were paired back with the same females in a 1:1 breeding scheme. The presence of a vaginal plug was checked each morning and used to time pregnancy. The presence of a vaginal plug was designated as gestational day (GD) 0.5 and female mice lacking vaginal plugs for 7 days were excluded. At GD 4.5, the males were removed from the breeding cage and the diet of the dam was switched as appropriate to either BFD, PB-FD, AFD or remained on NCD. Dams were maintained on their respective diets until weaning of the pups at postnatal day (P) 21. Litter size was adjusted to ensure adequate and standardized nutrition until weaning. Litters containing more than eight pups were culled down to eight pups at P1 and only these litters or those with six to eight pups naturally were used for future experiments. Offspring from a minimum of two separate litters per group were used for every experiment.
Paradigm of developmental exposure to BFD during gestation or lactation selectively
The exact same experimental paradigm as described above was followed with the differences that dams were either exposed exclusively to BFD from GD 4.5 until birth (BFDges; exposure during gestation only) or from birth (P0) until weaning (P21) (BFDlac; exposure during lactation only).
Paradigm of developmental exposure to fat-related sensory cues (‘caged BFD’)
For caged food cohorts, the accessible diet remained NCD for the entire duration but a wire mesh containing 100 g of either NCD or BFD was placed into the bottom of the food rack and replaced every other day. Breeding strategy and litter size controls were performed as detailed above.
Metabolic phenotyping: time lineMaternal metabolic phenotyping
Dams were weighed every 3 days during gestation and lactation. At weaning of the pups, body composition was analysed using micro-CT and mice were fasted overnight and blood was collected for HOMA-IR measurements. For LC–MS-based analysis of the hydrophobic (lipids fractions), 12 days after giving birth, milk was collected as described below as well as trunk blood upon killing.
Offspring metabolic phenotyping
Pups were weaned at P21 onto their respective diets and were weighed weekly throughout their lifespan. An insulin tolerance test (ITT) and body composition pre-HFD were performed at 6 and 8 weeks of age, respectively. At 8 weeks of age, animals from both groups were switched onto a HFD (post-HFD). At 12 weeks old (4 weeks on HFD), an ITT post-HFD was performed. At 16 weeks of age, resting blood glucose measurements were taken using a handheld glucometer from venous tail blood. At 18 weeks old, another micro-CT scan was performed for body composition analysis and body length measurements were taken from snout to anus using a ruler.
Offspring metabolic phenotyping (M71ReaChR)
Metabolic phenotyping of mice optogenetically stimulated during neonatal life was performed as described above but mice were fed an HFD enriched with acetophenone (0.25%).
Adult cohort metabolic phenotyping
Mice for adult cohorts were purchased from Charles River at 5–6 weeks old, housed four mice per cage and allowed to acclimate for 2–3 weeks in the animal facility. At 8 weeks old, mice were changed to BFD or maintained on NCD. Mice were then metabolically phenotyped according to a similar schedule to offspring mice. In short, at 16 weeks old, mice of both groups were switched onto HFD (HFDlard, post-HFDlard). TSE metabolic phenotyping cages were used during the change to HFD at 16 weeks old (pre- and post-HFD), an ITT post-HFD was performed at 20 weeks old, and resting blood glucose was taken at 24 weeks old.
Metabolic phenotyping: proceduresInsulin tolerance test
Each mouse underwent 1 week of habituation to daily handling and mock i.p. administration to minimize the stress induced by handling. Cages were changed the evening before the ITT to provide consistently clean cages and minimize leftover food spilling into the bedding. ITTs were performed in ad libitum-fed mice 2 h into the light cycle and food was withdrawn from the cage at the beginning of each test. Following determination of body weights and basal blood glucose concentrations, mice received an i.p. injection of 0.75 U kg−1 body weight of human insulin (Insuman Rapid, Sanofi Aventis) dissolved in room temperature 0.9% saline (Berlin Chemie). Blood glucose concentrations were measured from venous tail blood at baseline, 15, 30 and 60 min after injection using an automatic glucose monitor (Contour Ascensia, Bayer HealthCare).
Body composition analysis
Body composition analysis was performed using micro-computed tomography (micro-CT)-based imaging of mice under isoflurane anaesthesia. Data acquisition was performed in an IVIS Spectrum CT-scanner (Caliper LifeScience) using the IVIS LivingImage Software v.4.3.1. Quantification of fat and lean mass was performed with a modification of the Vinci software package v.4.61.038. Lean and fat mass were normalized to 100% before statistical analysis.
ELISA
Circulating leptin, CCK and PYY levels were measured in ad libitum-fed dams 5 h into the light phase. Circulating insulin levels were measured in overnight-fasted mice. For refeeding experiments, circulating leptin, CCK and PYY levels were measured in 12 weeks old overnight-fasted female mice. To perform experiments in HFDlard non-naive mice, mice have received small pieces of HFDlard pellets at least three times before experiments. Blood was collected by decapitation 10 min after HFD feeding. The blood was then centrifuged for 10 min at 10,000g, and the serum fraction was collected and snap frozen in liquid nitrogen. Insulin ELISA (Crystal Chem, 90080), leptin ELISA (Crystal Chem, 90030), PYY ELISA (Crystal Chem, 81501) and CCK ELISA (RayBiotech, EIA-CCK) were performed according to the manufacturer’s protocol.
HOMA-IR calculation
HOMA-IR was calculated as insulin (µIU ml−1) multiplied per glucose (mmol l−1) divided by 22.5.
Metabolic phenotyping cages
Measure of indirect calorimetry (EE and respiratory exchange ratio), locomotion and food intake were performed using the PhenoMaster System (TSE Systems). Mice were moved into training cages 3 days before data acquisition for acclimatation. Mice were maintained at 22 °C in sealed cages with ad libitum access to food and water. After 1 day in the PhenoMaster System to further acclimate, data acquisition was performed for 2 consecutive days starting 2 h into the light cycle on mice fed their respective diet (pre-HFDlard). Following the 2 days of pre-HFDlard at 3 h into light cycle, mice were switched onto HFDlard and recordings continued uninterrupted (post-HFDlard). All parameters were measured continuously and simultaneously. Data regarding EE were corrected for lean mass.
Milk collection
At 12 days after giving birth, lactating mothers were injected with 2IU oxytocin (Merck, O4375), anaesthetized under isoflurane (2–3%), and maintained at 37.0 ± 0.5 °C by a thermostatically controlled water heating system. Then, 5 min later the nipples were manually massaged to promote milk ejection, which was collected manually using a pipette. The milk was immediately snap frozen in liquid nitrogen for storage at –80 °C until analysis. Dams were killed after milk collection and tissue collected from offspring.
Amniotic fluid collection
Timed-pregnant dams carrying E15.5 fetuses fed NCD or BFD as described above were anaesthetized using 2% isoflurane and maintained on 2% isoflurane during the procedure. The abdominal skin was sterilized with Octenisept (Schülke) and an abdominal incision was made. The uterine horns were gently placed outside the abdominal cavity, sectioned and placed in phosphate-buffered saline (PBS) solution. Individual amniotic sacs were exposed and placed in a dry, sterile Petri dish. The amniotic membrane was carefully opened, allowing amniotic fluid to leak into the Petri dish, which was collected using a filtered micropipette and snap frozen in liquid nitrogen for storage at –80 °C until analysis. Following amniotic fluid collection, fetal and placental weight were recorded. Dams were killed after amniotic fluid collection and tissue collected from offspring.
Maternal behaviour
Maternal behaviour was assessed at P5. Dams were transported in their home cage to the test room 1 h before the experiment for acclimatization and remained in their home cage throughout the behavioural test. Trials were conducted between 4–7 h into the light phase. A camera (C920s HD Pro Webcam camera, Logitech) was mounted to capture a top-down view and connected to a laptop for recording. Each dam completed six trials, alternating between male and female pups (three males and three females). For each trial, one pup along with a small amount of bedding was placed on a heating pad. Once the dam returned to the nest, the pup was placed at the diagonally furthest point from the nest. Each trial lasted a maximum of 2 min. Analysis was conducted by measuring the time (in sec) required to retrieve the pup to the nest.
Gene expression analysisGene expression analysis
Samples were collected and snap frozen in liquid nitrogen. messenger RNA was extracted from the tissue using the mirVana miRNA Isolation kit (Ambion) and cDNA was produced by reverse transcription (High-capacity cDNA Reverse Transcription kit, Applied Biosciences). qPCR was performed using TaqMan Universal PCR Master Mix (Thermo Fisher) with Taqman probes (Thermo Fisher) listed below on a QuantStudio 7 Flex Real-Time PCR System (Applied Biosystems). Gapdh (Mm99999915_g1) was used as a housekeeping gene unless otherwise stated and data were analysed using the ΔCt method.
Dam hypothalamus
At weaning of the pups (P21) ad libitum-fed dams were killed by decapitation. Taqman probes used: Agrp (Mm00475829_g1), Npy (Mm00445771_m1), Pomc (Mm00435874_m1), Cartpt (Mm04210469_m1), Sst (Mm00436671_m1), Hcrt (Mm01964030_s1), Crh (Mm01293920_s1), Trh (Mm01182425_g1), Prl (Mm00599949_m1), Oxt (Mm01329577_g1) and Gal (Mm00439056_m1).
iBAT
Adult females’ offspring ad libitum-fed were killed by decapitation after 1 week of HFDlard feeding. Taqman probes used were Ucp1 (Mm01244861_m1), Adrb3 (Mm00442669_m1), Cidea (Mm00432554_m1), Pparg (Mm01184322_m1) and Ppargc1 (Mm01208835_m1).
Tongue
Adult offspring were killed by decapitation. Taqman probes used were Cd36 (Mm00432403_m1), Ffar4 (Mm00725193_m1) and Plcb2 (Mm01338057_m1).
Gene expression analysis following neonatal optogenetic stimulation
Cre-negative and OMP-Cre-positive offspring heterozygous for ReaChR were light-stimulated or mock-stimulated once at P3 and returned to their home cage with their mother for 1 h until killing by decapitation. Taqman probes used were Egr1 (Mm00656724_m1), Sema7a (Mm00441361_m1), Pcdh10 (Mm00477987_s1), Cnga4 (Mm01278645_m1), Neurog1 (Mm00440466_s1), Adcy3 (Mm00460371_m1) and Omp (Mm00448081_s1). Omp was used as a housekeeping gene.
HFD odour-induced hepatic p-mTOROlfactory exposure in freely behaving mice
Mice were split into cages containing two mice each 1 week before the experiment. A closed perforated metal tea ball was placed into their cages 3 days before to reduce the impact of a novel object during the experiment. Mice were acclimated to the experimental rooms and daily handling to minimize the effects of the odours related to the experimental room and the experimenter. After the acclimatization period, mice were fasted overnight and 2 h into the light cycle exposed to an empty tea ball (control, no odour) or to three pellets of HFDlard in a tea ball that allowed mice to smell HFDlard odours without physical access to the diet. Then, 30 min later, the mice were decapitated and the liver was quickly dissected and snap frozen in liquid nitrogen.
Protein isolation and western blotting
Approximately 50 mg liver tissue was transferred to a 2-ml tube containing 1 ml lysis buffer (containing Tris-HCl 25 mM, NaCl 25 mM, NP-40 1%, EDTA 1 mM, pH 7.4) with freshly added Halt Protease and Phosphatase Inhibitor Single-Use Cocktail (Thermo Fisher Scientific) and about 20 ZiO2 beads (VWR, CAS 432-0356). Samples were shaken for 2 min or until the tissue was fully digested. Samples were then centrifuged for 20 min at 15,000g and supernatant taken for protein quantification using Pierce BCA assay (Thermo Fisher Scientific). The protein samples were diluted to 2 mg ml−1 and mixed 1:4 with Laemmli sample buffer containing 10% β-mercaptoethanol.
Protein samples were run on a precast gel from Bio-Rad (4–15%) with 26 wells. Gels were run with a constant current of 130 V for 80 min. Transfer of gels to PVDF membranes was performed using Trans-Blot Turbo System (Bio-Rad) at 2.5 A and 25 V for 13 min. Gels were run in running buffer containing 25 mM Tris-HCl, 190 mM glycine and 0.1% SDS. Blots were blocked in 5% Western Blocking Solution (Roche, 11829200) in TBS-T (20 mM Tris, 150 mM NaCl, 0.1% Tween-20, pH 7.4) for 1 h at room temperature. Blots were incubated in the same solution containing anti-phospho mTOR (S2448) (Cell Signalling, 2971S) overnight at 4 °C. The next morning, the membrane was washed 3 × 5 min in TBS-T and the secondary HRP-conjugated anti-rabbit IgG (Invitrogen, 31466) was incubated at 1:1,000 for 60 min. Proteins were imaged using the Fusion Solo Vilber Lourmat system. Afterwards, blots were stripped in stripping buffer (10% SDS, 0.5 M Tris, 3.5% β-mercaptoethanol, pH 6.8) for 30 min at 56 °C and membranes were washed with TBS-T for 3 × 10 min and re-blocked. Incubation with anti-mTOR (Cell Signalling, 2972S) and HRP-conjugated anti-rabbit IgG (Invitrogen, 31466) was repeated as before. Band density was measured with ImageJ (National Institutes of Health). Data are presented as p-mTOR/mTOR and normalized to the average ratio of the non-food odour-exposed animals.
iBAT temperatureiBAT temperature upon food odour exposure
At 8 weeks old, an implantable electronic transponder with thermal couple (IPTT-300, Bio Medic Data Systems) was placed between the scapulae through a small incision under anaesthesia (2% isoflurane). The incision was allowed to heal for 1 week during which the animals were acclimated to handling and having temperature measurements taken with a wireless reader (DAS-8007C, Bio Medic Data Systems). The animals remained group housed. An empty tea ball was placed in the cages 3 days before the experimental day to eliminate effects of a novel object in the cage. On the day of experimentation, food was removed 2 h before the dark cycle. Baseline temperature measurements were taken every 20 min starting at the beginning of the dark cycle. Three readings for each mouse were averaged for the baseline value. A tea ball containing three pellets of HFDlard was then placed into the cage 1 h into dark cycle and temperature readings were taken 20 min later. All readings were recorded using DASHost 8000 software (Bio Medic Data Systems).
Longitudinal measurements of iBAT temperature
The 6–9-week-old mice were acclimatized to handling and thermal imaging procedure for approximately 2–3 weeks before experiments. Mice were carefully imaged using a thermal camera (FLIR E6, Teledyne FLIR LLC) by placing the camera at approximately 10 cm from the interscapular region. Temperature readings were taken weekly with an initial baseline measurement (pre-HFD; 9–12 weeks old), followed 1 week later by a diet change to HFDlard (1 h measurement) and 4-week follow-up measurements. Thermal images were analysed manually using thermal imaging software (FLIR Thermal Studio v.1.9.23.0).
Neonatal non-invasive optogenetic stimulation
OMP-Cre or M71-IRES-Cre were crossed to R26-LSL-ReaChR-mCitrine to allow for the expression of the optogenetic channelrhodopsin in all OSNs or only in OSNs expressing M71, respectively. The breeding strategy was similar to the one described above except that all dams were fed NCD. To optogenetically stimulate developing OSNs, a fibre optic cable (200 µm diameter, 0.48 NA, Doric Lenses) was held directly above but not in contact with the snout of the pup to deliver light at 625 nm (LEDFLS_625_625LED Fibre Light source, Doric Lenses). The stimulation paradigm was rectangular pulses of 100 ms ON/100 ms OFF for 3 s, 12 s OFF, repeated 12 times at 250 mA. This protocol has been previously demonstrated to replicate activity-dependent wiring of olfactory circuits and the glomerular map52. OMPReaChR mice and their Cre-negative control littermates were stimulated once at P3 for gene expression analysis. M71ReaChR and their Cre-negative control littermates were stimulated daily from P1 to P7. To minimize the stress induced by the manipulation, the experimenter’s gloves were rubbed with the bedding of the home cage to carry home cage and maternal odorants, and neonatal optogenetic stimulation was performed on a warm heating pad.
Neonatal non-invasive optogenetic stimulation coupled with calorie ingestion
The same paradigm as described above for neonatal non-invasive optogenetic stimulation was followed, with the modification that the pups were fed an Ensure liquid diet (Abbott, 640587) during stimulation. The Ensure diet was freshly prepared each day and warmed to 37 °C before being offered to the pups. 100 μl of Ensure was placed on a heating pad and pups were gently placed and kept close to it during light stimulation. All pups consumed Ensure during the experiments.
Neonatal optogenetic activation induced pS6 activation
Mice were optogenetically stimulated following the same paradigm described above. Mice were stimulated at P5 and then returned to the cage with the dam. At 1 h later, P5 pups were killed by decapitation. Heads, were quickly rinsed with 0.1 M PBS, followed by fixation in 4% PFA for 24 h. Then, they were transferred into 20% sucrose in PBS for 48 h, followed by being embedded in tissue freezing medium (14020108926, Leica Biosystems). After being frozen they were cut at 30 μm on a cryostat (Leica). For the immunostaining, slides were blocked with 5% donkey serum (diluted in 0.3% Triton and 1× PBS) for 1 h at room temperature, and then incubated with the primary antibody anti-GFP (1:500 dilution, ab13970, Abcam) and anti-pS6 (Ser244, Ser247) (1:1,000 dilution, 44-923G, Thermo Fisher Scientific), followed by incubation in the secondary antibody anti-rabbit IgG (1:500 dilution, A11012, Thermo Fisher Scientific). Tissue was coverslipped with Vectashield mounting solution with DAPI (Biozol VEC-H-1200, Vector Laboratories). Images were acquired using a confocal microscope Leica STELLARIS (Leica Microsystems) with a ×40 objective.
Behavioural testsDiet preference tests
Adult C57BL/6NCrl cohorts
The 7-week-old C57BL/6NCrl mice were single housed for 1 week and acclimated to two food hoppers (TSE Systems) for 4 days. Following the acclimation period, baseline food intake was measured for 3 days. The baseline food intake depicted in the figure represents the average daily food intake during these 3 days. The following day, mice were switched on HFDlard versus BFD or NCD versus BFD shortly before the onset of the dark cycle. Food intake was monitored for 5 days. For representation, baseline NCD intake is plotted as the average of the 3 days of NCD measurements.
NCDdev and BFDdev cohorts
At 8 weeks old, mice were single housed for 1 week and acclimated to two food hoppers allowing manual weighing of food intake. Following the acclimation period, on day 1, the diets in the food hoppers were changed to one food hopper containing HFDlard and the other containing CD, novel for both groups. For three consecutive days, food intake was recorded once per day after the onset of the light cycle and converted to calories of diet consumed. Data were calculated as percentage of calories consumed from HFDlard over the control diet.
Longitudinal food intake measurements
At 7 weeks old, mice were single housed for 1 week to acclimate to the food hopper and water bottle of the PhenoMaster System (TSE Systems). Mice had ad libitum access to food and water at all times. At 8 weeks old, mice were switched to HFDlard and food intake was measured until 20 weeks of age. Food intake was measured daily for the first 2 weeks on HFD and biweekly from week 2–10 on HFD.
Latency to eat
At 6 weeks old, the latency to eat was measured in overnight-fasted mice to determine relative attraction or aversion to AFD. Approximately 3 h into the light cycle, mice were placed individually into a clean cage and allowed to acclimate for 2 min. A single pellet of either NCD or AFD was then placed into the cage and the time it took for the mouse to approach and take the first bite of the pellet was measured. Mice were exposed to NCD or AFD in two consecutive weeks. The latency to eat AFD was normalized to the latency to eat NCD.
Odour investigation test
Approximately 3 h into the light cycle, ad libitum-fed mice were placed into a clean cage with a closed lid and allowed to acclimate for 2 min. Cotton swabs were dipped in solutions containing either vehicle (10% dimethylsulfoxide (DMSO) in water) or progressively increasing concentrations of acetophenone. The cotton swab was inserted through the hole for the water bottle spout to not open the cage between trials. The time spent sniffing each concentration, defined as head tilted up towards the cotton swab performing active sniffing motions, was measured over 2 min. The relative ratio of time spent sniffing each concentration of acetophenone normalized to time spent sniffing the vehicle was quantified for each group.
Habituation-dishabituation test
Approximately 3 h into the light cycle, ad libitum-fed adult female mice were moved into the experimental room and individually placed into a clean cage with a closed lid and allowed to acclimate for approximately 15 min. Mice were exposed to a sequential presentation of different odours presented in a perforated metal tea ball: empty tea ball (control), NCD, 1% AFD, 0.25% AFD and HFDlard. Each odour is presented in three consecutive trials for a period of 2 min, with a 15–20-s intertrial time. The time spent sniffing each odour was defined as the head pointed up and active nose/whisker movement. Quantification was based on video analysis taken from the side view.
Stereotaxic surgeryDopamine and calcium fibre photometry
All surgeries were performed in 8 weeks old mice. Mice were anaesthetized using 2% isoflurane and placed in a stereotaxic apparatus (Kopf Instruments). Before exposing the skull, the skin was sterilized with Octenisept (Schülke) and Anesderm (Pierre Fabre) was applied to the incision site. A small hole was drilled above target sites for viral delivery and fibre implantation. A 0.5-µl Neuros Hamilton Syringe (Hamilton 65450-03) was used to inject the virus at a rate of 100 nl min−1 and left in place for 5 min before being slowly withdrawn.
For photometry recordings, AAV9.Syn.Flex.GCaMP6s.WPRE.SV40 (400 nl, a gift from D. Kim & GENIE Project, Addgene viral prep #100845-AAV9) or AAV5-CAG-dLight1.1 (400 nl, a gift from L. Tian, Addgene viral prep #111067-AAV5) viruses were injected in the ARH and LAcbSh, respectively, using the following coordinates relative to bregma (in mm): ARH: −1.45 AP, 0.25 ML, −5.75 and −5.85 DV; LAcbSh: 1.0 AP, 1.75 ML, −4.6 and −4.7 DV. After virus delivery, an optical fibre (400 µm diameter, 0.48 NA, Doric Lenses) was implanted directly above the site of injection and secured using dental acrylic (Super-Bond C&B, Sun Medical). For postoperative care, mice were administered buprenorphine (0.1 mg kg−1, i.p.) and meloxicam (5 mg kg−1, subcutaneously) and received tramadol in the drinking water (1 mg ml−1) for 2 days before and 3 days after surgery.
M71ReaChR
adult optogenetic stimulation
The 12-week-old M71-IRES-Cre:R26-LSL-ReaChR-mCitrine mice were anaesthetized, placed in the stereotaxic apparatus and subsequently implanted with an optical fibre (200 µm diameter, 0.48 NA, Doric Lenses) targeting the right M71 glomeruli in the olfactory bulb, using the following coordinates relative to bregma (in mm): 4.1 AP, 0.25 ML and 1.0 DV.
Dual-colour fibre photometryData collection
Data were acquired using a RZ5P lock-digital processor controlled by Synapse software (Tucker-Davis Technologies) as described previously84.
Recording: dopamine sensor
Fibre photometry recording started 5 weeks after surgery to allow for mice recovery and optimal viral expression. Here, 2 weeks after surgery, mice were acclimated to the patch cord in the relevant behavioural setup for about 15 min each day. Mice were fasted overnight once during the fourth week post-surgery to reduce the future effects of first-time fasting. During the fifth week post-surgery, mice were fasted overnight and placed into a small experimental box 2–4 h into the light cycle. The experimental box (interior dimensions, 11.5 × 14 ×12 cm) is made in transparent plexiglass (wall thickness: 0.9 cm) with a circular cutout in the top corner (2 cm diameter) and a sliding lid containing a small hole to allow for the fibre optic cable to pass through. The experimental box was placed in a much larger arena with opaque black walls to block any view of the experimenter outside. A tube ran from outside the larger arena and through the circular cutout of the smaller box to allow for pellets to be delivered without interference from the experimenter. Food pellets were approximately ~20 mg each. After 10 min of acclimation, small pellets of BFD were provided to the mice at random intervals between 1 and 3 min to prevent anticipatory responses. An additional pellet was not given if the mouse had not eaten an already present pellet or if the mouse sat just below the delivery tube. In total, five pellets were delivered, as further pellets showed diminished responses during testing. Mice with no dopaminergic responses to refeeding on BFD were excluded from future experiments and analysis. This paradigm was repeated with 1 week in between for NCD and HFDlard. Mice were provided with a small pellet of HFDlard a few days before the trial to eliminate artifacts related to the novelty-induced neophobia.
Recording: calcium recording of AgRP neurons
Fibre photometry recording started 4 weeks after surgery to allow for mouse recovery and optimal viral expression. Mice were kept on their respective diet (pre-HFDlard) until 15 weeks of age and then both groups were switched to HFDlard (post-HFDlard). Starting 2 weeks after surgery, mice were acclimated to the patch cord in the relevant behavioural setup for about 20 min each day. At 3–4 weeks post-surgery, mice were placed into a clean cage without bedding and allowed to acclimate for 10 min. Mice were then injected i.p. with ghrelin (50 μg; Tocris 1465) diluted in 0.9% saline and the signal was recorded for 5 min. Mice with no changes in 465 nm fluorescence signal in response to ghrelin were excluded from future studies.
AgRP neuronal response to diets
At 5 weeks post-surgery, mice were then fasted overnight and placed in the photometry recording setup at 2–4 h into the light cycle. Following a 10 min baseline recording, a pellet of either NCD or HFDlard was placed into the cage and signal was recorded for 5 min. At 7 weeks post-surgery (at 15 weeks old), mice were switched to HFDlard in their home cage (post-HFDlard). At 3 weeks post-HFDlard, mice were fasted and recordings were repeated with NCD exposure. 1 week later, overnight fasting was repeated and recording was performed during HFDlard exposure.
AgRP neuronal response to hormones
AgRP neuronal fibre photometry recordings started 4 weeks post-surgery (after 1 week of acclimatation to the cable and behavioural setup). For all recording of AgRP activity dynamics in response to hormones mice were tested every week following the same experimental design: mice were placed in the behavioural setup and allowed to acclimate for 10 min before i.p. injection of a hormone and the signal was recorded for 20 min. At 12 weeks (4 weeks post-surgery), mice were injected i.p. with ghrelin (2 mg kg−1 BW; Tocris 1465). At 13 weeks, mice were fasted overnight, placed in the recording setup and injected i.p. with PYY (0.1 mg kg−1 BW; R&D Systems). After the recording session, animals were returned to their home cages and given ad libitum access to food. At 14 and 15 weeks, mice were fasted overnight and tested as described above for serotonin hydrochloride (2 mg kg−1 BW; Sigma-Aldrich) and glucagon (2 mg kg−1 BW; Bachem) respectively. All hormones were diluted in 0.9% saline and injected with a volume of 10 µl g−1 BW.
Viral expression and fibre placements
Virus expression and fibre placement were verified as described previously85.
Fibre photometry data analysis
All fibre photometry data analyses were performed using a custom MATLAB script. In the event of technical issues encountered on a recording day, such as malfunctions with the photometry rig during the recording session, the animal was excluded from this experimental time point.
dLight analysis
The initial 10 s of recording (laser power-up) was discarded and the data were downsampled at 50 Hz. To correct for artifactual signal fluctuations, the 405 nm Ca2+-independent isosbestic reference was aligned to the 465 nm signal using least-squares linear fit and then used as a baseline to compute the ΔF/F = (465 nm – scaled 405 nm)/(scaled 405 nm). The peri-event trace was extracted using a window spanning −30 s to +60 s around the moment of approach to the pellet manually identified from video recordings. Each trial was then z-scored using the −30 s to −20 s window as baseline to calculate the mean and s.d. All trials of a single animal and single diet were averaged, and the mean (z-scored) trace between 0 s to +5 s and +5 s to +10 s were taken to compare groups using unpaired t-test.
AgRP photometry analysis
The initial 10 s of recording was discarded and the data were smoothed using a 1-s moving-average window before being downsampled at 50 Hz. Peri-event traces were extracted from a window spanning −60 s to +180 s around the food pellet introduction or −120 s to +180 s around ghrelin injection. To avoid spurious influences of the stimulus administration, the baseline was defined as the window from −60 s to −20 s for food pellets and from −120 s to −60 s for CCK injection. For analysis of AgRP neuronal responses to ghrelin, PYY, 5-HT and glucagon, a similar approach was used with a smoothed using a 3-s moving-average window and downsampled at 50 Hz. Data were extracted from a window of −300 s to +1,200 s around the moment of injection. Baseline was defined from the window of −300 s to −60 s. The 465 nm signal was then z-scored, using this baseline to calculate the mean and s.d. Each trial was then z-scored using this baseline to calculate the mean and s.d. The mean (z-scored) trace between +15 s and +180 s was computed for each animal and then used to compare groups via unpaired t-tests.
M71 photostimulation-generated odour preference
The experiment followed the protocol for M72 photostimulation-generated odour preference described by Vetere et al.86, with minor modifications.
Odorant preparation
An M71-activating odorant (acetophenone; A10701, Sigma-Aldrich) or a non-M71-activating odorant (isoamyl acetate; 101231, Merck) were diluted in mineral oil (330779, Sigma-Aldrich) achieving a 40% concentration. The mixtures were stored in light-protected vials. To use the odorants, 50 μl of an odorant was pipetted onto filter paper on an inverted Petri dish (60 × 15 mm) and covered with cage bedding.
M71-glomerulus photostimulation protocol
The delivery of light pulses (625 nm, 85 mA, 4 Hz, 100 ms ON, 150 ms OFF) was designed to approximately resemble mice’s average natural duration of a sniff (100 ms) and breathing frequency (3–5 Hz)87,88,89,90,91,92.
Behavioural paradigm
All mice were naive to HFDlard, sucrose, acetophenone and isoamyl acetate before the experiment. The behavioural paradigm consisted of three phases: pre-exposure (day 1), optogenetic conditioning (day 2) and odour preference test (day 3). The experiment took place in a custom-designed preference test chamber made of white Plexiglas (15 × 40 × 25 cm) divided into two identical compartments by removable walls. On the first day, mice were plugged to the patch cord and underwent a 25-min pre-exposure to the test chamber, with each chamber compartment containing a non-scented Petri dish filled with bedding material. Mice did not receive any photostimulation during this first day. Then, mice were fasted overnight (16 h). On the second day, mice were exposed to a 25-min conditioning period. After the removable walls of the Plexiglas chamber were taken out, fasted mice were plugged to the patch cord and exposed to a non-scented Petri dish located in the centre of the chamber. The bedding in this Petri dish contained small pieces of HFDlard coated with 40% sucrose, which had been prepared the previous day. All mice engaged with the food pellets and ingested them. Simultaneously, mice received M71 photostimulation constantly during the 25 min of the conditioning, as described above. On the third day, the removable walls were re-inserted, and a Petri dish scented with either acetophenone or isoamyl acetate was placed into one of the chamber compartments, the location of which was randomly assigned. Mice were allowed to freely explore both compartments for 10 min without receiving any photostimulation. Mouse behaviour was continuously monitored with an overhead camera. These results reflect the whole 10-min observation period. Odour preference during the test day was calculated using the following formula: percentage of time spent in the acetophenone compartment – percentage of time spent in the isoamyl acetate compartment. After the experiments, mice were deeply anaesthetized with ketamine/xylazine and perfused transcardially with 0.1 M PBS followed by 4% PFA (pH 7.4) for future processing to verify fibre placements.
HistologyTissue processing
For fibre photometry experiments, mice were deeply anaesthetized with ketamine/xylazine and transcardially perfused with 4% PFA in 0.1 M PBS, pH 7.4. Tissues were post-fixed overnight in the same PFA solution. Brain tissues from photometry experiments were cryoprotected in 20% sucrose in PBS and cut at 30 μm on a cryostat (Leica). Tissue was coverslipped with Vectashield mounting solution with DAPI (Biozol VEC-H-1200). Sections were imaged using only the endogenous fluorescence of GCaMP6s or dLight1.1. White adipose and liver tissues were post-fixed for 24 h, dehydrated in 30% sucrose and embedded in paraffin before cutting at 5 μm on a vibratome and directly mounted. Tissue was stained with hematoxylin (GHS132, Sigma-Aldrich) and eosin (HT110232, Sigma-Aldrich) after deparaffinization.
Microscopy
All tissue was imaged on a Zeiss Imager M2 microscope and AxioVision v.4.2 software (Carl Zeiss) at ×20 magnification.
Quantification
Adipocyte size was quantified using ImageJ software for a minimum of 500 adipocytes per mouse from five randomly selected areas.
Ribosome immunoprecipitations (ps6-ribotrap)Tissue preparation and ribosome immunoprecipitation
Phosphorylated ribosome pulldown for the identification of olfactory receptor activation was adapted from previous protocols31. Experiments were performed on males C57Bl6/NCrl from 8–12 weeks old of age. Following 1 h of odour exposure, the olfactory epithelium was dissected on ice in buffer containing 1×HBSS (Gibco, with Ca2+ and Mg2+), 2.5 mM HEPES (pH 7.4), 35 mM glucose, 100 μg ml−1 cycloheximide, 5 mM sodium fluoride, 1 mM sodium orthovanadate, 1 mM sodium pyrophosphate and 1 mM β-glycerophosphate. Four epithelia were pooled and processed as described previously85.
RNA sequencing
Due to the low amount of input material, RNA-seq libraries were prepared using the Ovation RNASeq System V2 and Illumina Nextera XT protocol, as previously described93, and sequenced on an Illumina HiSeq 4000 (2 × 75 bp). We applied the community-curated nfcore rnaseq analysis pipeline v.1.446. The gene-level quantification was carried out using Salmon v.0.14.147 using the reference genome GRCm38. To normalize the immuno-pulldown for each sample to its background, we divided the Salmon gene count per sample using the formula countpulldowncountinput. The differential gene expression analysis based on the normalized counts was carried out using the DESeq2 v.1.26. R package94. Gene Ontology term analysis was carried out using the clusterProfiler v.3.14.3 R package95. Olfactory receptors with a Padj < 0.001 compared with no odour samples were defined as significantly activated, as defined by Jiang et al.31.
Two-phase metabolite extraction of polar and lipophilic metabolites (dams milk, blood and food pellets)
For the extraction of total lipids, 50 µl milk/plasma were collected in 2-ml round bottom Eppendorf tubes. For the extraction of the snap frozen material, the milk/plasma samples were allowed to thaw on ice. Lipids were then extracted by adding 1 ml pre-cooled (−20 °C) extraction buffer containing internal standard as described previously96.
LC–HRMS-based analysis of dams milk and blood lipids
The dried lipid extracts were resuspended in 200 µl UPLC-grade acetonitrile:isopropanol (70:30 (v:v)) before analysing them on a UPLC connected to a Tribrid Orbitrap HRMS as described previously96.
The obtained Thermo .raw files were converted to .mzXML files using MSConvert from ProteoWizard software package (http://proteowizard.sourceforge.net)97.
Data pre-processing for untargeted lipidomics analysis
Data pre-processing was performed with MZmine v.2.340 (http://mzmine.github.io/)98. Converted mzXML files were imported and extracted ione chromatograms were generated using the ADAP algorithm and peak picking was performed using the local minimum search algorithm. Isotopes were grouped and only peaks with at least one isotope were kept for further processing. Detailed parameters can be found in Supplementary Table 5. Finally, aligned peak lists were exported to CSV files for statistical analysis. Tables were corrected for solvent peaks by a blank subtraction. Features were removed when average peak area of samples was less than 2× average peak area of blanks.
Data evaluation
For data evaluation, CSV files were imported in Perseus (http://www.biochem.mpg.de/5111810/perseus)99. Data were further filtered for features that had a coverage of at least 70% valid data in at least one group. Filtered data were log2 transformed and missing values were imputed from normal distribution using the default settings.
LC–MS analysis of lipid extracts from food pellets
Lipidomics of food pellets was performed using a Thermo Vanquish Flex with a quaternary pump (Thermo Fisher Scientific) coupled to a TimTOF Pro2 mass spectrometer equipped with a heated ESI source (VIP-HESI) (Bruker Daltonics). Dried lipid samples were resuspended in 500 μl of acetronitril:isopropanol (70:30 (v:v)). The resuspended samples were cleared by a 5 min centrifugation at 16,000g and the 200 µl of the supernatants were transferred to 2-ml glass vials with 300 µl glass inserts (Chromatography Zubehör Trott). Then, 1 µl was injected in the UHPLC system and separated as described above. Compounds were detected in the mass spectrometer using data-dependent parallel accumulation serial fragmentation (DDA-PASEF) acquisition mode. All analysis were run in positive ionization mode using SA capillary voltage set to 4,500 V with an end plate off set of 500 V. Nebulizer pressure was set to 2 bar, dry gas was running with 8 l min−1 and a temperature of 230 °C. Sheet gas was running with 4 l min−1 and a temperature of 400 °C. For MS2 experiments masses were isolated with a width of 2 mD and fragmentation was induced with a collision energy of 30 eV.
LC–HRMS-based analysis of polar and semi-polar compounds
The stored (−80 °C) polar and semi-polar extracts were resuspended in 100 µl of a UPLC-grade water: methanol (50:50 (v:v)). From each analytical sample a volume of 20 µl was taken and pooled together. These pools were used as instrumental and sample stability quality controls (QCs), which are run after every tenth analytical sample in the sample sequence or after each replicate group. All samples and pools were placed in an Vanquish flex UHPLC (Thermo Fisher Scientific), which was connected to a TimsTOF Pro 2 HRMS, (Bruker Daltonics). Of each sample 1 µl was injected onto a 100 × 2.1 mm HSS T3 UPLC column, packed with 1.7-µm particles (Waters). The flow rate of the UHPLC was set to 500 μl min−1 and the buffer system consisted of buffer A (0.1% formic acid in UPLC-grade water) and buffer B (0.1% formic acid in UPLC-grade acetonitrile). The UHPLC gradient was as follows: 0–1 min 100% A (curve = 6), 1–11.5 min 100–0% A (curve = 7), 11.5–12 min 0% A, 12.0–12.1 min 0–100% A, 12.1–15 min 100% A. This leads to a total runtime of 15 min per sample. For better annotation some quality pool sample injections were run at the end of the sequence with a data-dependent parallel accumulation serial fragmentation (DDA-PASEF) acquisition mode with the same stepped mobility ramp to generate a MS2 spectra collection. Source parameters were as follows. Capillary voltage was set to 4,500 V with an end plate off set of 500 V. Nebulizer pressure was set to 2 bar, dry gas was running with 8 l min−1 and a temperature of 230 °C. Sheet gas was running with 4 l min−1 and a temperature of 400 °C. For MS2 experiments masses were isolated with a width of 2 mD and fragmentation was induced with a collision energy of 20 and 50 eV.
Feature extraction and data analysis
All samples were analysed in a randomized run-order after the column was conditioned with several blank and some QC sample injections. Before and after the set of samples pooled QC sample were injected. Lipidomics analysis and untargeted reversed phase analysis of food pellets were both performed with the following criteria: Raw data was further processed with MetaboScape (v.2024b) to generate an untargeted feature table. Detailed processing parameters can be found in Supplementary Table 3. Filtered feature table were normalized to the TIC median and further submitted to MetaboAnalyst (v.6.0). Missing values were replaced by LoDs (one-fifth of the minimum positive value of each variable). Remaining features (lipidomics 4084; untargeted RP 4020) were log-transformed and pareto scaled before further analysis.
Semi-targeted liquid chromatography-high-resolution mass spectrometry-based analysis of amine-containing metabolites
The LC–HRMS analysis of amine-containing compounds was performed using a QE-Plus high-resolution mass spectrometer coupled to a Vanquish UHPLC chromatography system (Thermo Fisher Scientific). Dried sample extracts were resuspended in 150 µl LC–MS-grade water for 10 min at 4 °C in a shaker at 1,500 rpm. After centrifugation, 50 µl of the extracts were mixed with 25 µl 100 mM sodium carbonate (Sigma), followed by the addition of 25 µl 2% (v/v) benzoylchloride (Sigma) in acetonitrile (UPC/MS-grade, Biosove), as reported previously. The derivatized samples were thoroughly mixed and kept at a temperature of 20 °C until LC–MS analysis96.
The LC–MS data analysis was performed using the open-source software MZmine 2 using MSConvert (2) (v.3.0.22060, Proteowizard). Compounds were annotated with an in-house library with an m/z tolerance of <5 ppm.
VOC analysis
Analysis was performed in the laboratories of GC–ToFMS Sensenet (Odournet).
Sample preparationDiets
Approximately 4 g of diet were introduced into an individual microchamber and heated to 30 °C. A thermodesorption tube (Tenax/Carbocarph) was inserted in the microchamber to collect a total volume of 1,000 ml of headspace.
Milk and amniotic fluid
A total of 1 μl of milk or amniotic fluid was directly spiked in a desorption tube (Tenax). The milk and amniotic fluid samples used for the analyses consisted of a pool of 4–5 dams.
Analytical methods
Combined thermal-desorption gas chromatography and time-of-flight mass spectrometry TD-GC–ToFMS system was used to generate a full quantitative scan of VOCs from the previously collected samples. Analyses were performed using a thermal desorption unit (Unity, Markes International), a gas chromatographer (7890, Agilent), a time-of-flight mass spectrometer (BenchTOF-dx model, Almsco) and a mid-polar DB-624 column was used for chromatographic separation (60 m, 250 μm, 1.4 μm; Agilent). Desorption tubes were heated to 300 °C with a helium flow rate of 50 ml min l−1 for 10 min (first desorption stage). Desorbed analytes were directed to a hydrophobic general purpose cold trap (10 °C, thermoelectric cooling), filled with Tenax TA and graphitized carbon. After flash-heating of the cold trap to 320 °C during 5 min (second desorption stage), analytes were injected into the chromatographic column for further separation, which took about 53 min. Molecules reaching the ToFMS detector were fragmented by electron impact ionization at 70 eV at a mass range of 28–330 amu. Deuterated toluene-d8 (Sigma-Aldrich) was used as an external standard for quantification. This compound was injected (10 ng) into an independent thermodesorption tube and was analysed by following strictly the same methodology as with the samples. Given the sensitivity of the method, two unused thermodesorption tubes were analysed as blanks, to exclude any potential contamination arising from these materials during the analysis. The deconvolution process for the chemical identification of the VOCs that were present in each analysed sample was carried out with the software TargetView v.3 (ALMSCO International). This algorithm identified the compounds of the chromatogram automatically based on an updated version of the NIST20 library. Chemical identifications were confirmed with at least 80% certainty. Results of each diet/sample were normalized as per cent for representation.
PET imaging
PET imaging was performed using an Inveon preclinical PET/CT system (Siemens) as previously described100.
Olfactory exposure during PET imaging
Olfactory exposure was performed as described85.
PET analysis
The kinetic model and statistical analysis was performed as previously described85,100.
Statistical analysis
Statistical analysis was performed with GraphPad Prism 8 unless otherwise stated. Student’s t-test, Wilcoxon test, multiple t-test, one-way analysis of variance (ANOVA), two-way ANOVA and three-way ANOVA were used when applicable. Data not representing a Gaussian distribution were analysed by nonparametric tests. Statistical data are represented in the figure legends. In brief, datasets comparing experiments in the same animal subjected to two different treatments were analysed for statistical significance using paired two-tailed Student’s t-test. Datasets with only two independent groups were analysed for statistical significance using unpaired two-tailed Student’s t-test. Datasets with more than two groups were analysed using a one-way ANOVA followed by a multiple-comparisons test. Datasets influenced by two independent factors were analysed using a two-way repeated-measures ANOVA, followed by Sidak’s or Tukey’s multiple-comparisons test or a mixed-effects model followed by a Tukey’s multiple-comparisons test in cases where data were missing at certain time points. Datasets subjected to two independent factors were analysed using two-way repeated-measures ANOVA followed by a Sidak’s multiple-comparisons test in case of missing values at some time points. Three-way (type III) ANOVAs were performed in R v.4.3.0. For between-subject designs, we used linear models fitted with the stats package and analysed with the car package; for repeated measures, we implemented linear mixed-effect models using the lmerTest package and its built-in ANOVA function. No statistical methods were used to predetermine sample sizes but our sample sizes are similar to those reported in previous publications. Data distribution was assumed to be normal but this was not formally tested. Data collection and analysis were not performed blind to the conditions of the experiments. Alpha was defined as 0.05 and significance was depicted by *P < 0.05, **P < 0.01 and ***P < 0.001. Code used for the analyses is available upon request.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.