
Clinical findings
The patient is the second child of healthy, non-consanguineous parents, who first presented at the age of 15 month in our neuropediatric outpatient clinic.
After an uneventful pregnancy and normal birth, two urinary tract infections in the second and fourth month of life occurred. A renal ultrasound was conducted and a duplex kidney with an enlarged kidney pelvis (right side) was diagnosed. A double kidney, also known as a double collecting system, is a congenital malformation in which, in the mildest form, only the renal pelvis is divided into two parts and, in the most severe form, one kidney has developed two complete urinary tracts. This is a relatively common variation of the urinary tract, occurring in about 1% of the population. Further renal examinations showed mild dilated ureter and mild disorders in the renal function (protein-crea-ratio).
The parents reported a motor developmental delay, showing crawling at 13 months of age, to stand up at 15 months of age, and to walk independently at 22 months of age. Particularly in the first year of life, proximal muscular weakness in the upper arms and shoulder girdle was noticeable, which made it impossible for the child to crawl up. During physiotherapy, very mild contractures in the elbow joint were noticed for the first time around 10 months of age. The cognitive development and language development were unremarkable.
Clinical examinations at the age of 15 months in our department of pediatric neurology (University Medicine Essen, Germany) revealed joint contractures of both elbows/ saddle thumb joint, muscular hypotonia of the trunk and atrophic muscles of the upper extremities with muscular weakness and macrocephaly. The elbow contractures are more limiting to function than weakness. Laboratory analysis presented mild elevated CK-level of max. 244 U/l (normal value < 180 U/l). At the age of 24 month, MRI of cranium, spine and shoulder girdle muscles including the upper arm muscles showed no abnormalities (data not shown). Due to the EDMD-like phenotype, developmental delay and mild elevated CK-level a muscle biopsy was performed for diagnostic purposes. At this time, the genetic diagnosis of NPS had not been made. Certainly, a muscle biopsy is not necessary to confirm an NPS.
Further neuropediatric follow-up examinations revealed constant muscle weakness of the upper extremities, scapulae alatae and mild motor developmental delays with myalgias (Fig. 1A). The index finger of the left hand showed a triangular lunula but no dystrophic signs (Fig. 1B). Physiotherapy and ergotherapy occur as supporting therapy—mild improvements of the contractures have been shown.
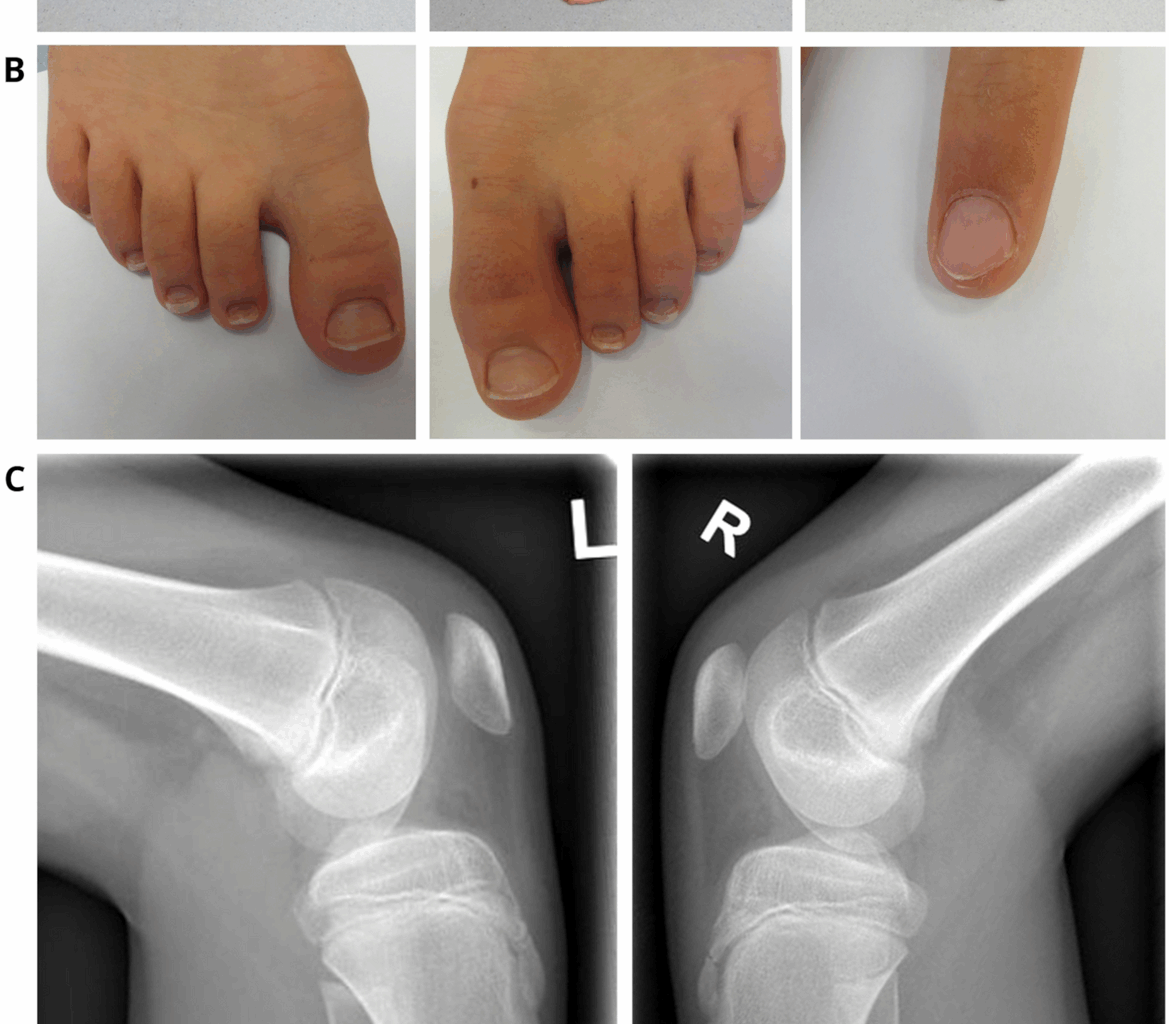
Fig. 1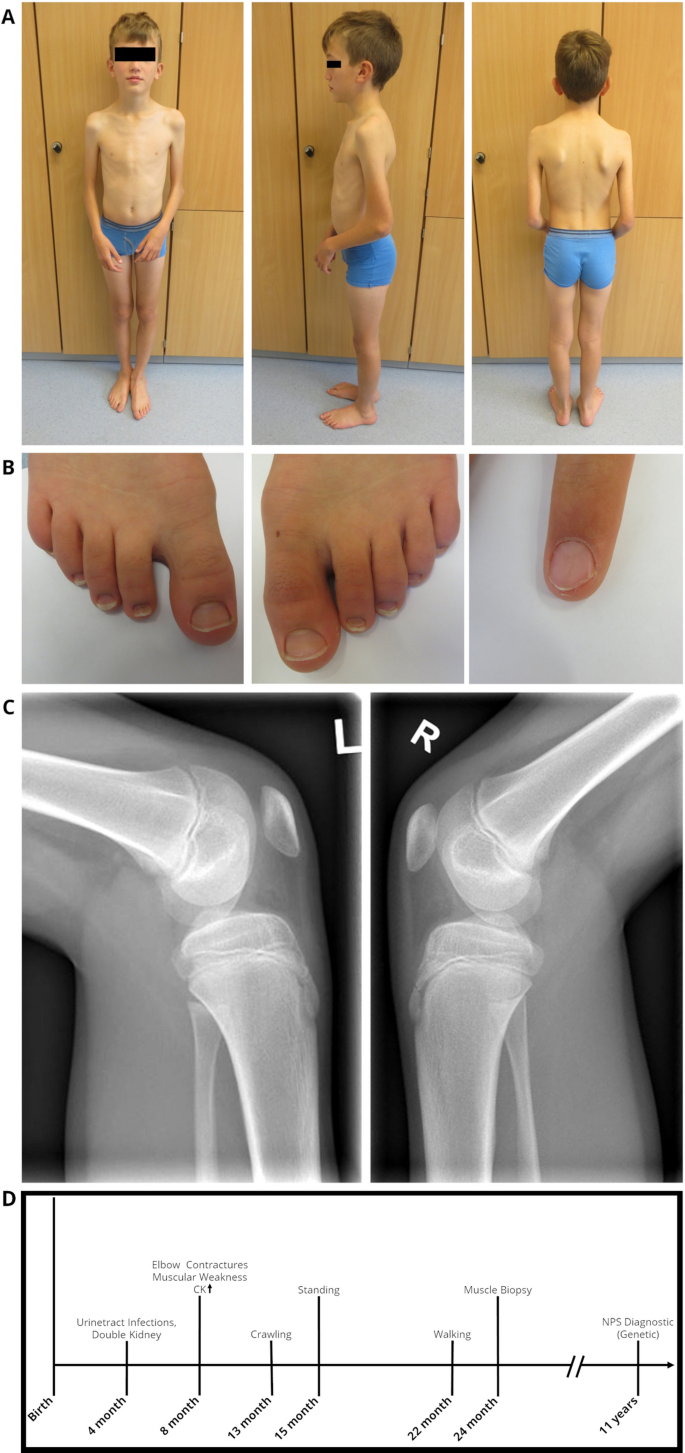
Clinical findings in our index. At the age of 12 years: A Pronounced elbow contractures on both sides, with atrophy of the upper arm muscles and scapulae alatae, B mild dystrophic nails of the feet and triangular lunula of the finger nail, C Patellar hypoplasia on both sides. D Schematic representation of the diagnostic work-up along with clinical findings in our NPS patient
Currently, at the age of 16, the patient is part of a football team and shows no muscle weakness, although the contractures of the elbow are more pronounced than at the time of the muscle biopsy.
Muscle biopsy findings
Since the patient developed symptoms mainly in the upper extremity resembling a EDMD-like phenotype, a biopsy of the deltoid muscle was collected for further diagnostic work-up at the age of 2 years and histology showed a skeletal musculature with very mild abnormalities defined by discrete atrophic fibres. The following routine stains were carried out on the basis of clinical suspicion of congenital myopathy, and an immunoblot analysis was performed for suspected EDMD. Protein abundance and distribution for Caveolin 3, Dystrophin (1/2/3), α-/β-/γ-/δ- Dystroglycan, Laminin α2, Emerin, Dysferlin, Collagen IV as well as Telethoin, Calpain, Dysferlin and LMX1B studied by immunofluorescence appeared normal (data not shown). Results of immunoblot of Emerin were unremarkable (data not shown). Microscopic studies of semi-thin sections (Fig. 2B) as well as ultra-structural investigations were performed to further elucidate potential morphological perturbations. Here, resin sections revealed only a moderate variation of fibre calibres (Fig. 2D). Ultrastructural analysis revealed marginal nuclei normal in shape and structure including integrity of the nuclear envelope. The sarcomere architecture was regular without specific alterations (Fig. 2F).
Fig. 2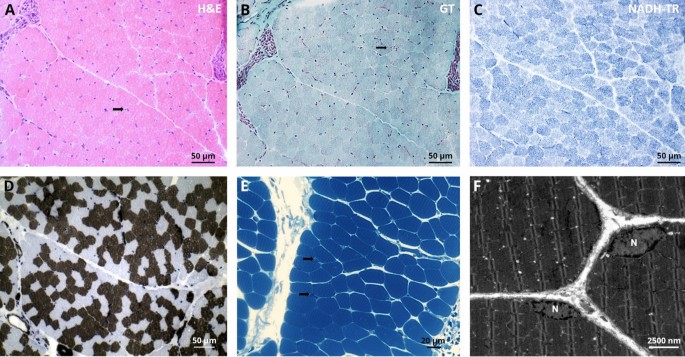
Histopathological findings in deltoid muscle of our NPS patient. Light microscopic findings on LMX1B mutant muscle, H&E stains and semi-thin sections show normal morphology with some atrophic fibres (arrow)(A + E). Gomori trichrome (GT) and nicotinamide adenine dinucleotide tetrazolium reductase (NADH-TR) show normal morphology. ATPase stain (pH 4.3) shows moderate variation of fibre calibres D. Ultrastructural analysis revealed marginal nuclei (N) normal in shape and structure including integrity of the nuclear envelope. The sarcomere architecture is regular without specific alterations F
Molecular genetic findings
Genetic diagnostic testing of the LMNA and COL6A1-3 genes revealed no pathological findings. Variants in ITGA7 gene (c.8246G > A; p.(Arg275His)) and RYR1 gene (c.2648C > T; p.(Ala882Val)) of uncertain significance were found. However, as those were also not fitting with the phenotype observed in our patient, the patient was included in a research project (NMD-GPS; https://nmd-gps.net/) for exome sequencing unveiling a heterozygous missense variant in LMX1B (c.668G > A; p.(Arg223Gln)) in exon 4 (https://www.ncbi.nlm.nih.gov/clinvar/RCV000007422/). Sanger-based segregation studies confirmed a de novo origin of the variant.
Proteomic Profiling
To analyse the potential impact on the biochemical integrity of skeletal muscle, unbiased proteomic profiling was conducted in a data-independent-acquisition mode. This analytical approach allowed the quantification of proteins spanning eight orders of magnitude (Fig. 3A) and identified 128 upregulated and 64 downregulated proteins (Fig. 3B). A proteomaps-based pathway analysis was conducted for increased and decreased proteins separately: Upregulated proteins impact on various biological processes including activation of the complement system, metabolism, RNA-transport as well as exosome function and endocytosis. Downregulated proteins also impact on exosome function in addition to ribosomal function, tRNA-loading, MAPK signaling, and protein ubiquitination among others (Fig. 3C).
Fig. 3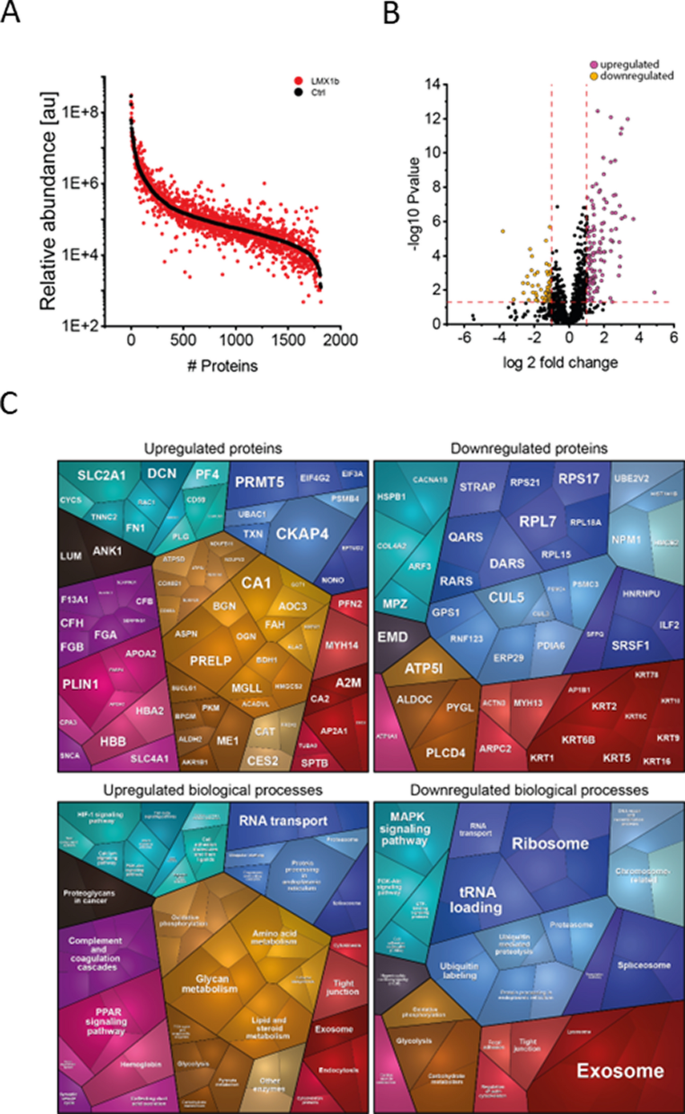
Proteomic findings in deltoid muscle of our NPS patient. A Abundance plot showing the dynamic range of all proteins identified in proteins extracts of deltoid muscle via liquid chromatography coupled to tandem mass spectrometry based on their relative quantification using always the 3 highest abundant peptides for each protein, allowing protein comparison within an experiment. All identified proteins of the controls (black) are sorted with decreasing abundance while the patient (red) was plotted in the same order to directly compare the different abundances. All identified proteins cover a dynamic range of eight orders of magnitude. B Volcano plot highlighting statistically significant increased proteins (purple dots) as well as decreased proteins (yellow dots). C Proteomaps-based in silico analysis of cellular functions affected by the respective dysregulated proteins. Dysregulated proteins are shown in the upper panel separated for upregulated proteins (left) and downregulated proteins (right). Affected biological processes are shown in the lower panel accordingly
Among the 192 differential abundant proteins, 16 of the up-regulated and 26 of the downregulated proteins are localized in the nucleus (Table A1). Of note, 59 proteins are associated with diseases and some proteins are associated with diseases that phenotypically overlap with the spectrum of clinical presentation seen in NPS (Table 2). These disease associations of dysregulated proteins for instance include lipodystrophy (PLN1), deafness (MYH14) and peripheral neuropathy with myopathy, hoarseness and hearing loss (MYH14). GTR1 is associated with neurologic disorders and GLUT1 deficiency syndrome is clinically characterized by encephalopathy, delayed development, microcephaly, motor incoordination, and spasticity, Epilepsy, dystonia and other neurological defects syndrome.
Transcriptomic profiling
Finally, we aimed to address the unmet question if LXM1B is expressed in human skeletal muscle. For this purpose, we utilized a transcriptomic dataset consisting of bulk RNA-sequencing of eight non-diseased controls. We compared the expression of LXM1B to genes with known expression in skeletal muscle, including COL1A2 and TTN. Here, we observed that LMX1B is not expressed in human muscle cells (Fig. 4).
Fig. 4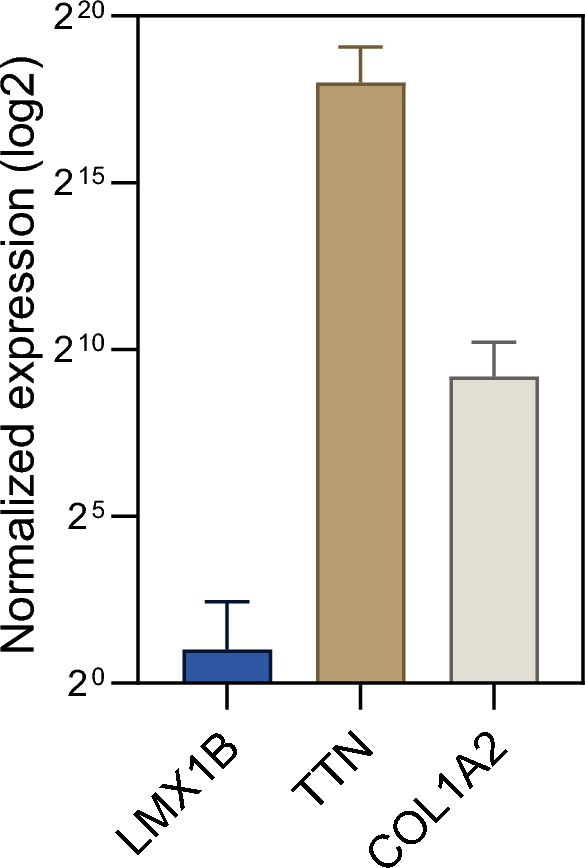
Transcript studies toward expression analysis of LMX1B in human muscle. Bulk RNA-sequencing of eight non-diseased controls. We compared the expression of LXM1B to genes with known expression in skeletal muscle (COL1A2 and TTN). LMX1B is not expressed in muscle tissue