
Experimental design
The design of our evolution experiments has been described in detail previously [25]. Briefly, using Davis Minimal Medium, E. coli JA122 was evolved in triplicate in aerobic, glucose-limited (0.0125% w/v) chemostats at a fixed dilution rate of 0.2 h−1 and at a constant temperature of 30 °C. Relative to E. coli K12 MG1655 our founder strain JA122 has an elevated mutation rate (1.0 × 10−7 vs 3.6 × 10−9 bp generation) due to a nonsense mutation in base excision repair glycosylase, MutY (L299*). JA122 also contains nonsense mutations in the housekeeping (σD aka σ70 (RpoD), E26*) and stationary phase (σS aka σ38 (RpoS), Q33*) sigma factors, each of which positions ribonucleic (RNA) polymerase holoenzyme to its respective consensus sequence (note: after a sigma factor’s first mention, we hereafter refer to it by its gene name). However, the founder strain also carries a nonsense suppressor tRNA capable of suppressing all three types of nonsense mutations [32, 33]. Thus, while the ancestral MutY defect increases mutational load on our populations, the presence of a glnX suppressor softens the effect of nonsense mutations. Suppressor activity may even be enhanced by the slow growth conditions [33, 34] imposed by resource limitation.
Identification of high-value targets and mutation clusters
Here, we analyzed a collection of functional modules whose components become targets of selection when E. coli evolves under continuous glucose limitation. Targets of selection were defined as genes and regulatory elements in which the number of observed mutations exceeds the number that would be expected by random chance, given the observed number of mutations and gene/element sizes (Table 1; [25]). Thirty-nine targets meeting a 5% false-discovery rate (FDR) threshold were further examined for evidence of non-random patterns of mutation either in their primary sequence or in their 3-dimensional structures, the latter using ClusterExplorer [35], the nonrandom mutations cluster (NMC) algorithm [36], and the Identification of Protein Amino Acid Clustering (iPAC) program [37]. Nineteen protein coding genes/intergenic regions exhibited at least one significant cluster of mutations (Table 1). Many of these clusters precisely define intergenic regulatory regions or occur in known protein structural elements, such as domains involved in catalysis, protein-RNA, or protein–protein interactions. In targets where de novo mutations were not clustered but still present in excess, they often occurred in regions that could alter protein activity or regulation in ways that enhance acquisition or utilization of the limiting nutrient, facilitate energy conservation, or increase cells’ residence time in the chemostat. A majority of genes deemed to be high-value targets of selection fell into one of four functional categories: regulatory proteins (galS, malT, malK, rho, hfq, proQ, ompR, rpoS, rpoA, gatZ), proteins that act in lipopolysaccharide export (lptA, lptB, lptC, lptD, lptG), multifunctional inner membrane proteins (opgG, opgH), and proteins required to construct cell surface appendages (fliG, fliH, fliP, fimH). Within each of these categories we found examples of groups of genes whose products collaborate in specific biological processes, qualifying them to be regarded as components of a functional module (Fig. 1).
Table 1 Protein coding genes/intergenic regions that had more mutations than expected by chanceFig. 1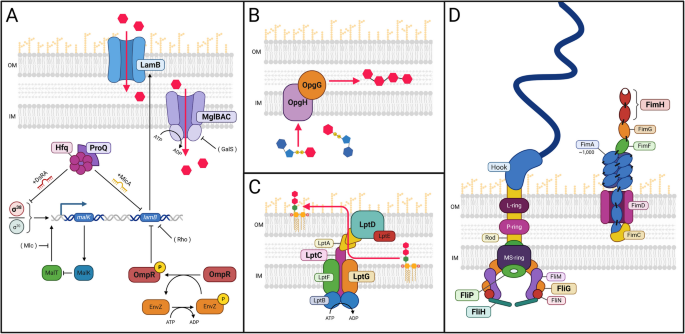
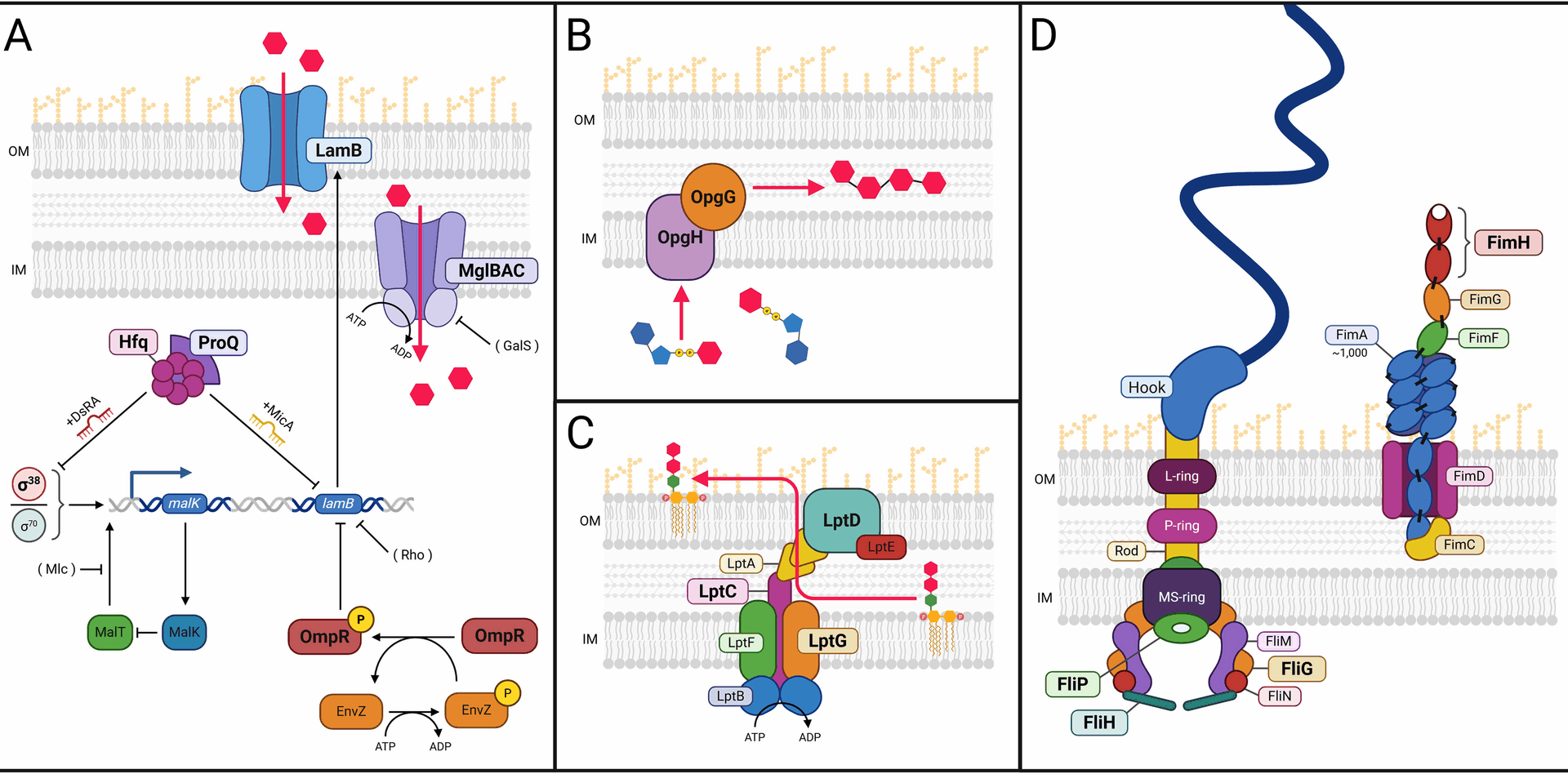
Functional modules whose genetic components are mutated more often than expected by chance in E. coli evolved under glucose limitation. A Glucose assimilation under glucose limitation. In glucose-limited chemostats, glucose diffusion across the outer membrane into the periplasmic space is facilitated by glycoporin LamB, and its transport from the periplasmic space into the cytoplasm occurs via the inner membrane transport complex MglBAC. LamB expression is regulated by RNA chaperones Hfq and ProQ, DNA-binding protein OmpR, and transcriptional regulators, RpoD (σ70) and RpoS (σ38). B OPG biosynthesis. Transport of cytoplasmic UDP-glucose and its assembly into osmoregulated periplasmic glucans (OPGs) requires OpgG and OpgH, which are frequently mutated in our experiments. C LPS trafficking. Transport and secretion of lipopolysaccharide (LPS) to the outer cell surface requires the Lpt complex, elements of which are targets of selection. D Cell surface appendages. Proteins in functional modules required to construct appendages used in motility (flagellae, left) and attachment (fimbriae, right) are frequently mutated. (OM=Outer membrane, IM=Inner membrane, Magenta hexagons=Glucose, Boldface=Proteins discussed at length in the Results)
Regulatory proteinsUnder resource limitation, regulatory mutations that influence glucose uptake and conservation offer high-value targets for selection
Among the functional modules most frequently mutated in our experiments are those required to scavenge glucose when it is the limiting substrate. One such module is organized around the diffusion of glucose across the E. coli outer membrane; another is organized around transport of glucose across the inner membrane (Fig. 1A). When glucose is limiting E. coli access, this substrate via proteins associated with the movement of galactose and maltose [13, 25, 31, 32, 38,39,40,41], notably the high-affinity outer membrane maltose/glucose porin, LamB [42]. While no mutations were observed at the lamB locus itself, our data were enriched in mutations likely to alter the activity of four lamB effectors: malT, malK, rho, and hfq, all of which were enriched for clustered mutations (Table 1). Because each of these effectors represses lamB expression, either directly (e.g., rho) or indirectly (e.g., malK), and because all 87 de novo alleles are either nonsense or missense mutations in key functional domains, all likely de-repress lamB transcription ([25] and references therein), enhancing cells’ capacity to scavenge limiting glucose.
Another component of this functional module is built around regulating expression of the D-galactose/methyl-β-D-galactoside transporter MglBAC, which under glucose limitation moves glucose from the periplasmic space across the inner cell membrane [25, 43] (Fig. 1A). As with LamB, we observed no mutations in MglBAC uptake system itself. However, galS, a key effector of mglBAC expression, is the most frequently mutated gene in our population sequencing dataset, though these mutations show no evidence of clustering (Table 1). The DNA-binding protein GalS negatively regulates mglBAC transcription [44]. Because every new galS allele is either a nonsense or missense mutation, all could therefore be expected to diminish GalS deoxyribonucleic acid (DNA) binding affinity, resulting in mglBAC de-repression, which would enhance cells’ ability to assimilate limiting glucose. The spectrum and evolutionary dynamics of mutations arising in the functional modules depicted in Fig. 1A are discussed at length in our previous communication, as are the structural consequences of mutations in galS, malT, malK, and rho [25]. Below, we discuss the structural and functional consequences of non-random mutations in other lamB effectors, RNA chaperones hfq and proQ, response regulator ompR, RNA polymerase holoenzyme components, rpoA and rpoS, as well as in the putative protein chaperone gatZ.
RNA chaperone Hfq is a target of recurrent mutation under glucose limitation
The most frequently mutated gene in our population sequencing dataset, hfq, is also among the most frequently mutated genes in our clonal sequencing dataset and shows multiple mutational clusters ([25] and Table 1). All but one of the de novo hfq alleles are missense mutations, with examples of the same residue being repeatedly mutated within the same experimental population (e.g., Pro64Thr and Pro64Gln), or the same residues being mutated in all experimental populations (e.g., Arg17Leu, Gly29Cys) (Fig. 2A, Additional File 2: Table S1 and [25]). hfq encodes the RNA-binding protein Hfq, which regulates post-transcriptional small RNA (sRNA)/mRNA interactions that modulate cellular processes ranging from central metabolism and amino acid biosynthesis to peptidoglycan biosynthesis, motility, and cell division ([45,46,47] and refs therein). Hfq also acts as a general stress response regulator by interacting with mRNAs that encode alternative RNA polymerase sigma factors σ24 (RpoE), σ32 (RpoH), and RpoS, each of which can substitute for housekeeping sigma factor RpoD to produce RNA polymerase holoenzyme required to initiate transcription [48]. Each σ-factor controls the expression of a different set of genes by binding to specific consensus sequences −10 and −35 upstream of the transcriptional start site.
Fig. 2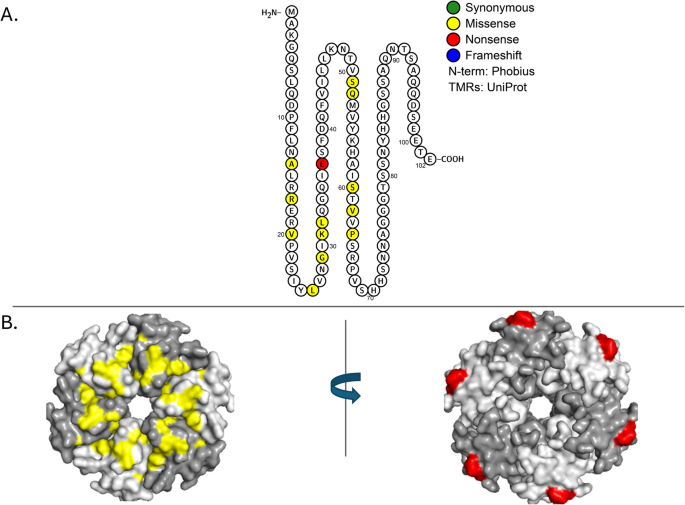
Mutations in RNA chaperone Hfq (N= 24, P=6.91E-40). A Location of mutations on the primary structure of the RNA-binding protein, Hfq. Residues reported to be involved in RNA binding include: Gln8, Phe39, Lys56, and His57 [53]. Mutations occurring at amino acid positions 17, 26, 29, 31, 32, 60, 62, and 64 were recorded in more than one chemostat. B Rotated 3-D image of the ancestral genotype with missense mutations located on the distal face (in residues 26, 29, 31, 32, 52, 60, 62 and 64) highlighted in yellow, and Arg17Leu (located on the rim) highlighted in red. Alternating subunits of Hfq are in different shades of grey
E. coli Hfq is a 102 amino-acid protein with a disordered N-terminal domain (aa 1–6), a core Sm-like domain (aa 7–65) and an unstructured C-terminal tail (aa 66–102) [49,50,51,52] (Fig. 2A). The active protein is composed of six monomers organized into a ring-like structure having three surface domains—the distal face, lateral face (or rim), and proximal face (Fig. 2B) [51, 53, 54]. The distal face of each Hfq monomer contains a tripartite motif with an adenine-binding groove (A-site), a purine interaction site (R-site) and a nonselective site (E-site) (Fig. 3B). This motif has strong affinity for (ARN)x nucleotide repeats frequently found in the 5′ UTR of mRNAs such as the rpoS mRNA leader [55]. Lateral face residues on Hfq help facilitate sRNA-mRNA annealing, while those on its proximal face bind AU-rich tail regions of sRNAs [56,57,58,59]. For example, Hfq promotes RpoS translation at suboptimal E. coli temperatures by facilitating interaction between rpoS mRNA on the distal face with DsrA bound to the proximal face, leading to exposure of an obscured rpoS ribosome binding site [59,60,61,62,63,64,65].
Fig. 3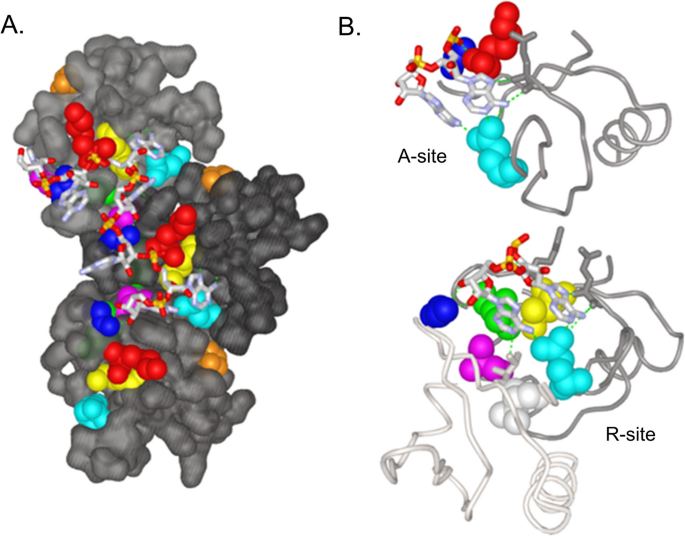
Hfq modulates transcription of stationary phase sigma factor (σs) by directly interacting with A-rich regions of the rpoS leader sequence. A Distal view surface representation of three Hfq subunits bound to A7 oligonucleotide representing the A-rich region of the rpoS mRNA leader. Unchanged residues are colored grey and residues affected by mutations are colored as follows: Leu26=green, Gly29=blue, Lys31=red, Leu32=yellow, Gln52=cyan, Ser60=pink, Val62=white, Pro64=orange. Hydrogen bonds (3.5 Å) between the A7 oligonucleotide and Hfq are depicted as green dashed lines. B Close up view of adenine nucleotides interacting with the A-site and R-site. Residues colored as in panel A
We observed a total of 24 mutations in hfq, 14 of which were unique (Table 1 and Additional File 2: Tables S1 and S2). Every unique mutation occurred within hfq’s core Sm-like domain. Most were concentrated on the distal face (Leu26Phe, Gly29Cys, Lys31Asn, Leu32Met, Gln52His, Ser60Tyr, Val62Phe, Pro64Thr, Pro64Gln), and some (Pro64Gln) arose independently multiple times (Fig. 3A, B and Additional File 2: Tables S1 and S2) [59, 66]. Specific changes at many of these residues (Gly29Cys, Lys31Asn, Leu32Met, Gln52His, and Ser60Tyr) (Fig. 2B, Fig. 3A) have previously been implicated in Hfq’s interaction with A-rich RNA molecules as well as with ADP and ATP [60]. A secondary RNA and ADP binding site is located on the rim of the Hfq hexamer and includes charged residues Arg16, Arg17, and Arg19 [67]. A mutation at Arg17 (Arg17Leu) arose independently in all three chemostats.
Because the majority of hfq mutations precisely delineate the binding site of the rpoS leader (A7 oligonucleotide), these mutations likely affect regulation of RpoS translation by the small non-coding RNA DsrA (Fig. 3A). Amino acid changes in, or adjacent to, the A- and R-sites of the ARN binding motif may disrupt the base stacking and hydrogen bond formation needed for Hfq to interact with the A-rich RpoS leader (Fig. 3B) [60]. Hfq mutations resulting in diminished translation of RpoS are likely to increase lamB transcription by reducing RpoS competition with RpoD for core RNA polymerase [20] (Fig. 1A). In addition, Hfq collaborates with antisense sRNA MicA to downregulate lamB expression [55]; disruption of this regulatory circuit is likely to increase levels of glycoporin LamB (Fig. 1A). Strikingly, no mutations were detected that altered residues either on the hexamer’s proximal face or in the C-terminal tail, suggesting there may adaptive constraints on Hfq evolution in this environment.
RNA chaperone ProQ is also repeatedly mutated under glucose limitation
ProQ was originally identified as a non-essential osmoregulatory factor required to optimally express proline channel protein ProP [68, 69]. ProQ was later shown to be a major RNA-binding regulatory protein that has both strand exchange and RNA duplexing activities [70]. Multiple RNA targets for ProQ have been proposed, with many sRNA species co-precipitating with this protein [71, 72]. Deuteration protection assays reveal specific regions where ProQ preferentially binds different sRNAs [73]. E. coli ProQ consists of an N-terminal domain, spanning residues 1–130, that is very similar to that of the ProQ paralog, FinO. This N-terminal domain is connected to a Tudor-like C-terminal domain (residues 180–232) by a 63 aa linker region. All three regions are proposed to bind RNA [73]. In the N-terminal domain seven positively charged residues (Arg32, Arg69, Arg80, Arg100, Lys101, Lys107, and Arg114 (colored in yellow in Fig. 4B) form a patch that is highly protected in deuteration protection assays; this patch has been implicated in binding to the 3′UTRs of sRNAs that include the late stationary phase ncRNA SraB [73, 74], DNA-damage inducible ncRNA SraB [75].
Fig. 4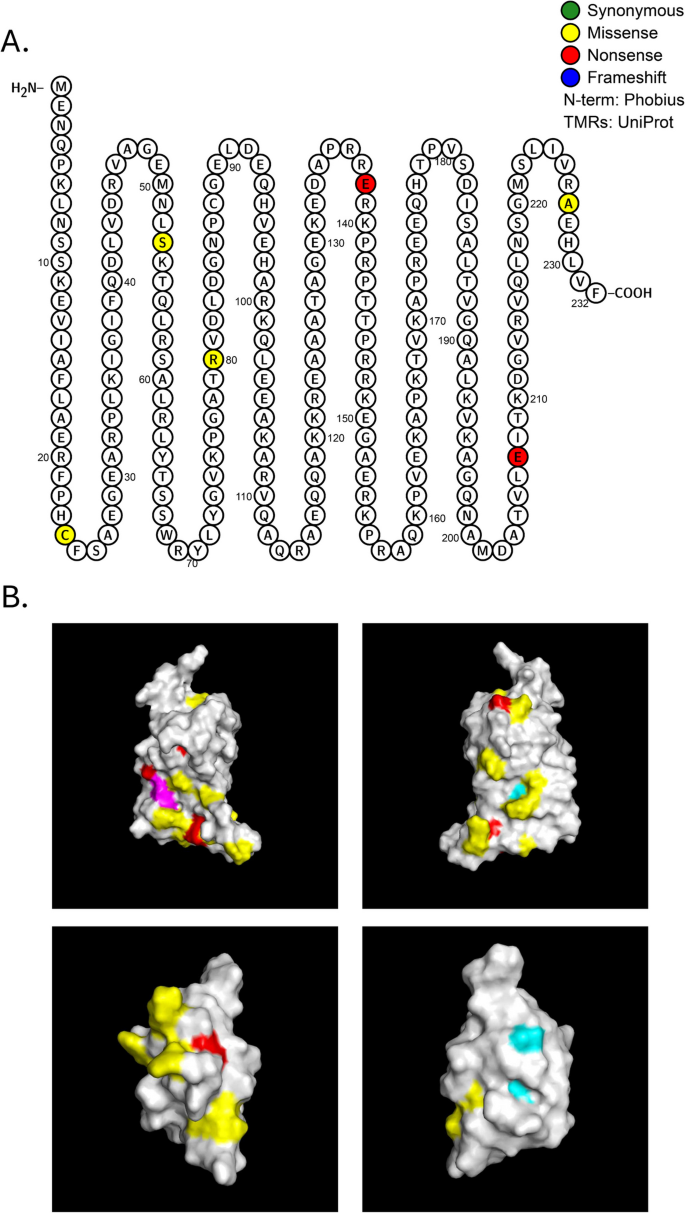
Mutations in RNA chaperone ProQ (N=6,
P=1.81E-05). A Protter diagram of ProQ protein. B Top: 3-dimensional model of ProQ N-terminal domain rotated 1800. Positively charged residues implicated in RNA binding (Arg32, Arg69, Arg80, Arg100, Lys101, Lys107, Arg 114) are indicated in yellow [73]. Mutations arising in our evolution experiments include Arg80Leu (magenta), Gly85Val and Leu103Pro in red (top left); on the opposite side of the N-terminal domain are these mutations: Ala106Glu and Ser53Ile (red) and Cys88* (cyan) (top right). Bottom: 3-dimensional model of ProQ C-terminal domain rotated 180°. Ala227Glu is adjacent to Arg226 in the proposed RNA binding patch (red; bottom left), while Gly189Val and Ala203Asp (cyan; bottom right) are on the opposite side of the molecule are not in the vicinity of any positively charged residues
Like hfq, the RNA chaperone proQ is mutated more often than expected by chance (Table 1, Fig. 4, and Additional File 1: Figure S1). While we did not detect significant clustering of proQ mutations, we did find in the N-terminal patch substitution of a polar for a non-polar amino acid (Arg80Leu) as well as two other missense mutations (Gly85Val and Leu103Pro) (colored in red, Fig. 4A). Three additional mutated residues are located on the other side of the N-terminal domain adjacent to other charged residues: Ala106Glu and Cys88*(Gln) in the vicinity of Lys75 and Arg109, and Ser53Ile adjacent to Lys54 (Fig. 4B, left). In ProQ’s C-terminal domain one de novo mutation (Ala227Glu) is located adjacent to Arg226 in a proposed RNA binding patch (red, Fig. 4B, right), while two other mutations (Gly189Val and Ala203Asp) on the opposite side of the molecule are not in the vicinity of any positively charged residues (cyan, Fig. 4B, right). All these mutations are likely to destabilize the ProQ protein.
It is perhaps not mere coincidence that RNA chaperones Hfq and ProQ are both mutated far more often than expected by chance. Recent co-immunoprecipitation and RIL-seq data indicate that each chaperone can bind hundreds of target sRNAs and mRNAs [76]. In most instances, the two proteins bind different targets, with ProQ showing marked preference for sRNAs, although more sRNAs overall are bound by Hfq [77]. In scores of cases, the RNA-RNA interactomes of the two chaperones overlap, setting up the potential for ProQ and Hfq to compete for the same target, as they do for rybB sRNA and, to a lesser extent, for micA sRNA [76, 78]. Expression of both these σE-dependent sRNAs [79] is known to modulate expression of stationary phase transcription factor σ38/RpoS ([80]; and Fig. 1A), whose diminished activity has been repeatedly associated with increased fitness among E. coli evolved under glucose limitation [25, 30, 39, 81]. Also, as noted above, micA RNA bound to Hfq acts as a post-transcriptional repressor of LamB synthesis (also see [82]). It is tempting to speculate that MicA bound to ProQ may open an alternative route to LamB repression, which, if mutationally blocked, would prove adaptive under glucose limitation.
DNA-binding dual transcriptional regulator OmpR is recurrently mutated
Outer membrane permeability is an important determinant of adaptation to very low concentrations of glucose [38]. When glucose is non-limiting, it can enter the periplasm passively through outer-membrane porins OmpC and OmpF. However, differences in the relative amounts of these porins have also been observed during glucose-limited chemostat growth [31, 40, 83]. The regulation of OmpC and OmpF is complex; their relative expression is controlled in part by the EnvZ/OmpR two-component regulatory system that responds to changes in medium osmolarity or pH [84, 85]. OmpR is active either as a dimer or monomer and is composed of an N-terminal receiver domain and a C-terminal DNA-binding effector domain that also interacts with the RNA polymerase α subunit, RpoA [86,87,88,89]. When phosphorylated, OmpR’s interaction with RpoA is favored, and ompF and ompC expression is active but modulated in a reciprocal manner: i.e., when external osmolarity is high, ompF expression is favored over that of ompC, whereas when osmolarity is low the reverse is true [89, 90]. Mutations that affect porin regulation have been reported in both domains of OmpR, as well as in its cognate histidine kinase EnvZ and in RNA polymerase α-subunit RpoA [89]. OmpR-P is also involved in regulating a number of other genes including lamB, malE, flagellar master operon genes flhDC, curli production genes csgDEFG, and the small regulatory RNAs micF, omrA, and omrB (Fig. 1A) [78, 91,92,93,94,95,96]. While OmpR is not considered essential, its inactivation or deletion in the presence of constitutive malT mutations is lethal due to outer membrane changes that stem from LamB hyperaccumulation [78, 97]. This effect can be mitigated by expression of the LptB component of the LPS transporter, establishing a link between mutation of OmpR and MalT, LamB overexpression, membrane stress, and LPS transport [78], all of which appear to play roles in E. coli’s evolutionary adaptation to chronic glucose limitation (Fig. 1).
Although no mutation clusters were detected in ompR, this locus was mutated more frequently than would be expected by chance (Table 1). Two of the five mutations observed (Lys6Asn and Ala35Asp) occurred in the N-terminal receiver domain (Fig. 5A) while the remaining three (Ser174Arg, Pro179Thr, and Leu228Met) were in the C-terminal effector domain (Fig. 5B). When mapped onto a model of the OmpR receiver domain, the Ala35Asp mutation is close to the Asp55 phosphorylation residue and the rest of the catalytic triad (Asp11 and Asp12), suggesting that it may interfere with phosphorylation or transmission of the phosphorylation signal to the effector domain (Fig. 5A). Based on the OmpR crystal structure [98], the three effector domain mutations (Ser174Arg, Pro179Thr, and Leu228Met) occur at the end of the α1 helix, in the loop between helices α1 and α2, and at the N-terminal end of sheet β5 (Fig. 5B, magenta/red). OmpR DNA-binding activity is known to involve residues in α3 (Fig. 5B,green), while OmpR-RpoA binding occurs in the loops between helices α1 and α2, in helix α2, and in the loop between helices α2 and α3 (Fig. 5B, blue/magenta) [99,100,101,102]. The relative location of mutations Ser174Arg at the end of helix α1, Pro179Thr between helices α1 and α2, and Leu228Met in sheet β5 suggest that these de novo mutations are more likely to impact OmpR association with RpoA than its binding to DNA. OmpR mutants carrying a Pro179Leu allele demonstrate a transcription negative phenotype, but still can interact with EnvZ and bind DNA, further supporting the hypothesis that mutation at this residue interferes with RpoA binding [101]. Remarkably, as we discuss below, not only is OmpR mutated where it interacts with RpoA, but RpoA is also frequently mutated where it binds to OmpR. The fitness benefit of such mutations is clear: Impaired OmpR function is known to result in constitutive expression of glycoporin LamB, the major route by limiting glucose is transported across the E. coli cell wall under glucose limiting conditions [78].
Fig. 5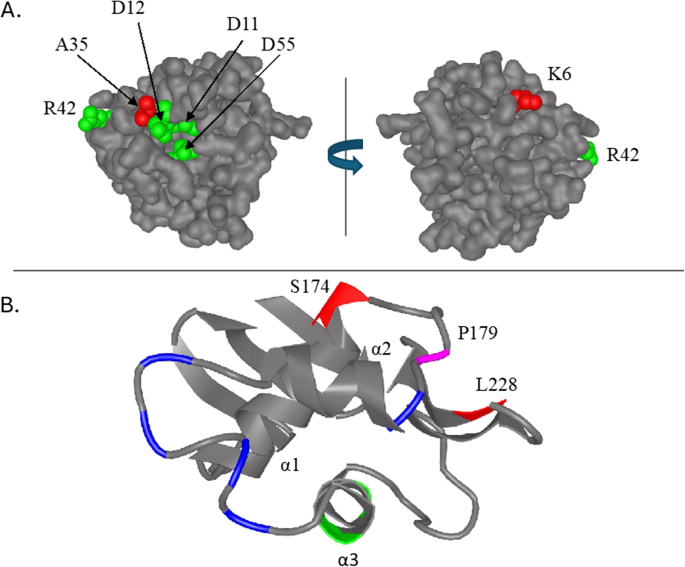
Location and frequency of mutations in the outer membrane protein regulator, OmpR (N=5, P=2.41E-4). A SWISSMODEL homology model of N-terminal receiver domain of OmpR based on the structure of YycF (PDB 2zwn, sequence identity = 47.06%). Residues that form the active site (Asp11 and Asp12 and Asp55) as well as a residue that, when mutated, affects transcriptional activation (R42) are shown in green. Residues that were mutated in this study (Lys6Asn and Ala35Asp) are colored red. B Ribbon diagram of the C-terminal effector domain of OmpR (aa 130-239, PDB 1OPC). Helix α3 contains DNA contact residues V203, R207 and R209 (green), which interact with thymine, guanine and the phosphate backbone in the major groove. OmpR and RNA polymerase α subunit (RpoA) interactions occur in the loop between helices α1 and α2, in helix α2 and in the loop between helices α2 and α3 (blue and magenta,respectively)
The α-subunit of RNA polymerase, RpoA, and stationary phase transcription factor, σ70/RpoS, are both recurrently mutated under glucose limitation
rpoA encodes the α-subunit of RNA polymerase, which consists of a C-terminal domain and an N-terminal domain connected by a flexible linker. Dimerization of RpoA, which is controlled by its carboxy-terminal domain, is required to assemble the RNA polymerase core complex [103]. rpoA was recurrently mutated in our replicate experiments (Table 1, Fig. 6, and Additional File 2: Table S1). One of targeted residues, Pro322, was hit three times, in each case causing a change from a non-polar to a polar residue (Pro322Thr in chemostat 1 and Pro322Thr/Pro322Gln in chemostat 2) and was therefore recognized as a significant cluster by the NMC and iPAC algorithms (Table 1). Mutations in the C-terminal half of RpoA affect the activity of many different positive RpoA regulators including CRP, FNR, and OmpR (reviewed in [104]). Rather than exhibiting a general inhibitory effect, most of these mutations appear to be specific to certain regulators and clustered in discrete patches along the RpoA primary sequence. For example, mutations in the C-terminal 10 amino acids, in particular aa322 and 323, prevent transcription of the OmpR-controlled genes ompF and ompC [89, 104]. Expression phenotype can depend on the nature of the amino acid substitution. Pro322Ser (nonpolar to polar) mutations reduce expression of both porin genes, whereas Pro323Leu (nonpolar to nonpolar) reduces ompC transcription but negligibly affects that of ompF [89]. Because all three RpoA mutations we observed at residue 322 changed proline to a nonpolar amino acid (leucine in one instance and threonine twice), we hypothesize that in lineages containing these mutations only ompC expression is affected.
Fig. 6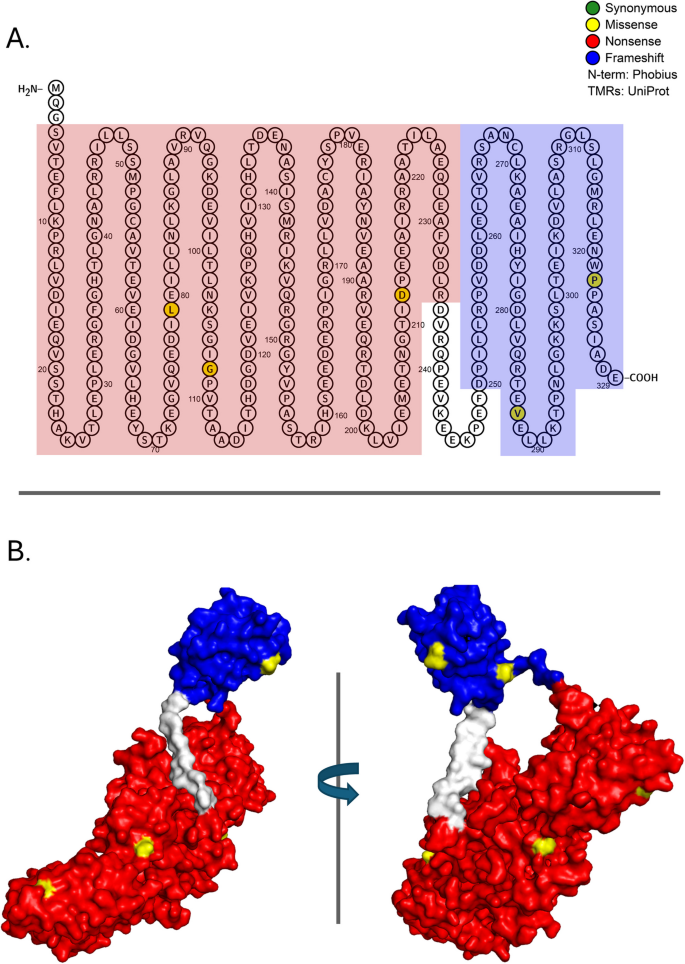
Mutations in proteins required for transcription initiation: a-subunit of the RNA polymerase core enzyme, RpoA (N=7, P=1.27E-05). RpoA consists of an alpha N-terminal domain that interacts with DNA-binding transcriptional dual regulator Crp at class II promoters plus an alpha C-terminal domain. A Location of mutations on the primary structure of the RpoA protein. Red and blue represent, respectively, functional domains 1 and 2. Amino acids that are filled represent a specific mutation that arose in the evolution experiments. B Rotated 3-D image of the ancestral protein with functional domains 1 and 2 colored in red and blue, respectively. Observed missense mutations are highlighted in yellow
As noted, the ancestral E. coli strain used for these evolutions contained several mutations likely to influence transcriptional regulation, in particular a nonsense mutation in housekeeping sigma factor, RpoD (Glu26*), a nonsense mutation in the stationary phase transcription factor, RpoS (Gln33*), and a nonsense suppressor mutation in glnX tRNA that suppresses amber, ochre, and opal mutations. Our three experimental populations accumulated an additional 9 rpoS mutations, far more than expected by chance (Table 1). Six of these were missense mutations, and two (Arg299Ser, Phe278Leu) were determined by iPAC to cluster significantly (Table 1, Fig. 7, and AdditionalFile 2: Table S1). Both mutations occurred in domain 4 (262–315) that encompasses the RpoS DNA-binding region (288–307) with its helix-turn-helix motif. Because housekeeping RpoD regulates expression of E. coli glucose scavenging proteins and because RpoS and RpoD compete for the core RNA polymerase (RNAP), the fitness advantage that accrues to RpoS mutants under glucose limitation has long been attributed to reduced competition between RpoD and RpoS for RNAP (e.g., [81]), which would enable glucose scavenging under slow growth conditions. As the ancestor in our experiments contains N-terminal nonsense mutations in each sigma factor as well as a tRNA suppressor mutation capable of bypassing both, it may be that excess mutations in RpoS represent one of several mechanisms that serve to minimize RpoD-RpoS competition, ensuring maximal expression of genes involved in glucose transport and assimilation (see Fig. 1A).
Fig. 7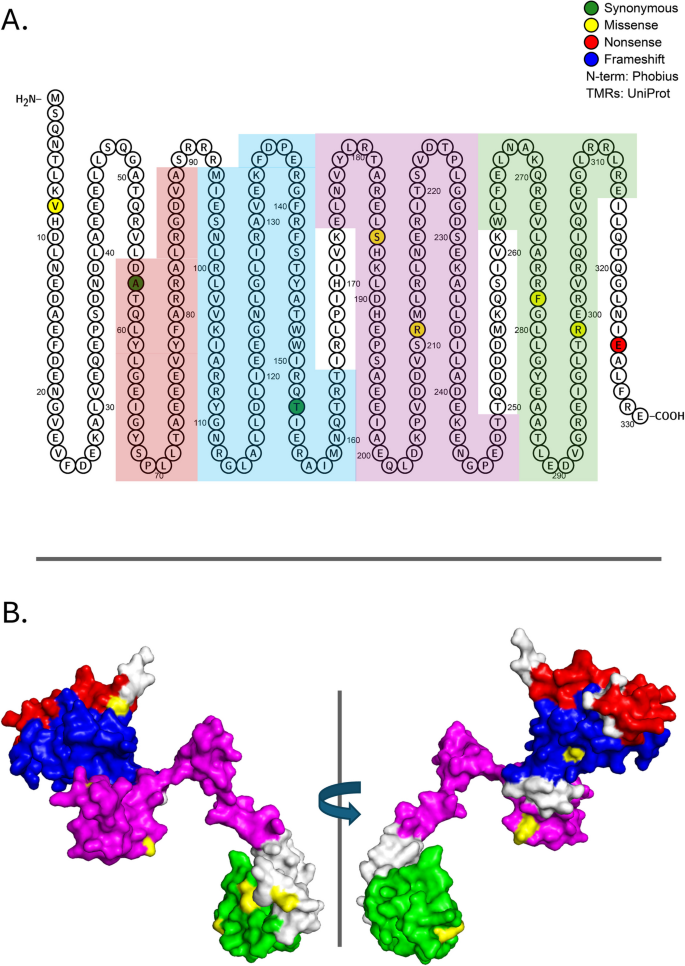
Mutations in proteins required for transcription initiation: RpoS (N=9, P=9.71E-05). RpoS consists of four sigma-70 factor domains (DT1–4) that function in DNA binding and melting and that interact with RNA polymerase subunits RpoA, RpoB, and RpoC. A Location of mutations on the primary structure of the RpoS protein. Red, blue, magenta, and green highlighting represent domains 1-4, respectively. Amino acids that are filled represent a specific mutation that arose in the evolution experiments. B Rotated 3-D image of the ancestral genotype with functional domains 1-4 colored inred, blue, magenta, and green, respectively. Observed missense mutations are highlighted in yellow
A post-translational regulatory protein becomes a target of selection under glucose limitation
gatZ was recurrently mutated in our experiments, though these mutations were not clustered (Table 1, Additional File 1: Figure S2). While GatZ appears to lack catalytic activity, evidence suggests that it acts as a protein chaperone to ensure proper folding of GatY, tagatose-1,6-bisphosphatase aldolase [105]. GatZ inactivation has been shown to be beneficial under anerobic conditions and conducive for H2 production from glycerol [106]. Interestingly, recurrent mutations inactivating the gat operon have been shown to be beneficial among E. coli experimentally evolved in gnotobiotic mice [107,108,109]. In multiple instances, adaptive mutations were either IS insertions (79%) or short deletions (21%) in the gatYZABCD operon, whose gene products collectively allow for galactitol catabolism. All these mutations exerted polar effects and produced the same phenotype: the inability to metabolize galactitol. All conferred a fitness advantage [108], perhaps related to dispensing with an unnecessary and costly pathway. In this regard, it is noteworthy that a majority of gatZ mutations were either nonsense mutations or indels and that we also observed nonsense mutations in gatC and gatY, in all a total of 13 mutations in the module defined by this single operon (AdditionalFile 2: Tables S1 and S2). Similar to [108], none of our mutant gat alleles ever went to fixation. On the other hand, none ever went extinct, and in two populations their final frequency was ~ 20%. While the role played by GatYZ under continuous glucose limitation has not been explored, it is noteworthy that these proteins reversibly interconvert D-tagatose 1,6-bisphosphate with the glycolytic intermediates D-glyceraldehyde 3-phosphate (G3P) and dihydroxyacetone phosphate (DHAP) [110]. Gat inactivation may therefore also serve as a mechanism to prevent diversion of limiting carbon to non-essential pathways. This hypothesis is consistent with fixation of a GatY nonsense mutation (G49*) in the predominant clone isolated from a previous E. coli evolution experiment performed under glucose limitation [32].
Multifunctional inner membrane proteinsProteins required for synthesis of periplasmic glucans are more frequently mutated than expected by chance
Osmoregulated periplasmic glucans (OPGs), formerly termed membrane-derived oligosaccharides (MDOs), consist of 5–24 subunits of D-glucose connected by β-glycosidic linkages. As their name implies, periplasmic concentrations of OPGs vary inversely with extracellular osmolarity. To date, OPGs have been reported in four of six major subdivisions of the Proteobacteria, indicating that they may be essential components of the cell envelope across this group [111]. In E. coli, OPGs consist of 5–12 glucose residues; cellular OPG content can range from as much as 5% total cell dry weight in low osmolarity medium to as little as 0.5% dry weight in high osmolarity medium. OPG biosynthesis requires OpgH, which spans the inner membrane, and OpgG, which closely interacts with OpgH in the periplasm (Fig. 1B). OPG molecules can be decorated with phosphoglycerol, succinyl, and phosphoethanolamine residues by the OpgB, OpgC, and OpgE/OpgD proteins, respectively, but it is OpgH/OpgG that transport UDP-glucose out of the cytoplasm and form b-glycosidic linkages between glucose monomers. Recent data suggest that these inner membrane proteins have functions other than transport and catalysis, including the coordination of cell division and cell size via the intermediate UDP-glucose [112].
In our evolution experiments opgH was much more frequently mutated than expected by chance, being the third most frequently mutated gene in our population and clone sequencing datasets (Table 1). OpgH mutations were found to be significantly clustered by the Cluster Explorer (positions 333–457) and NMC (positions 334–512, inclusive) algorithms, with 18 of 31 independent mutations occurring in this region of the primary sequence. OpgH is predicted to have three cytoplasmic regions connected by eight transmembrane domains that span the inner cell membrane (Fig. 8) [113]. Currently, no solved structure exists for displaying the location of OpgH amino acid substitutions in 3-dimensional space. Nevertheless, our understanding of the protein’s basic topology makes it clear that mutations cluster within the middle cytoplasmic region, a region that exhibits features reminiscent of glucosyltransferases; the C-terminal portion of this region has been shown to be essential for catalytic activity [113]. Thus, missense mutations here may compromise assembly of the OPG backbone. They may also compromise transport of UDP-glucose to the periplasm, which would also impair OPG synthesis, but not due to a catalytic defect. Also, for reasons that are poorly understood, OPG assembly requires acyl carrier protein (ACP). While ACP necessarily interacts with one or more of the three cytoplasmic regions, the exact site of this interaction is not currently known. It is noteworthy that while opgH is a frequent mutational target, only a handful of de novo opgH alleles ever surpass 10% frequency in our replicate evolutions (Fig. 8). The most successful of these, Pro434Thr, attained 5% frequency by 200 generations and eventually rose to 78% by the end of the experiment.
Fig. 8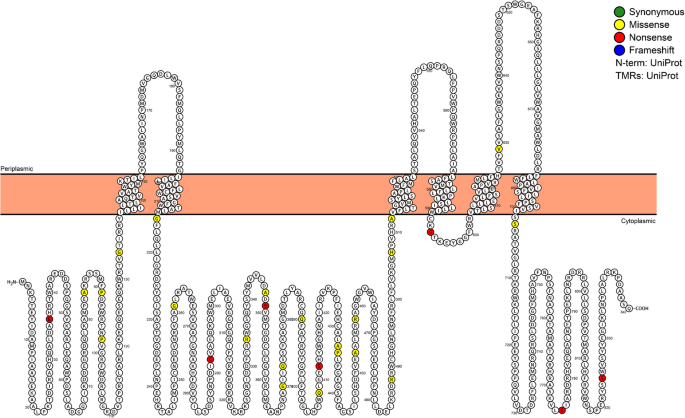
Mutations in proteins required for multifunctional inner membrane protein export: Osmoregulated periplasmic glucans protein, OpgH (N=31, P=8.74E-27). Location of de
novo mutations in the OpgH primary structure.Amino acids that are filled represent a specific mutation that arose in the evolution experiments. Amino acid positions 370, 373, and 408 were each mutated in multiple chemostats. OpgH is a transmembrane protein, and the inner membrane is represented by peach-colored bar
opgG is the first gene to be transcribed in the opgG/opgH operon and is OpgH’s periplasmic partner in the synthesis and placement of periplasmic glucans between the inner and outer membranes (Fig. 1B). Although opgG was also more frequently mutated than expected by chance (Table 1), no mutations were significantly clustered. All OpgG mutations were either missense or nonsense mutations, including 2 independent nonsense mutations at Glu81* (Fig. 9). While Glu147* eventually rose to 13% frequency, no other opgG allele exceeded 2.5% frequency across our replicate evolutions [25]. This outcome stands in contrast with a prior E. coli evolution experiment where an opgG nonsense mutation (E487*) became fixed in the predominant lineage isolated after 765 generations of continuous glucose limitation [32]. We also observed a novel allele in another inner membrane protein, opgC, which encodes for succinyl transferase. While this missense mutation (Gln64Lys) arose in a single population at 100 generations, it was one of only a handful of alleles across replicate experiments that ever went to fixation. Whether this allele was a driver or passenger mutation is not currently known.
Fig. 9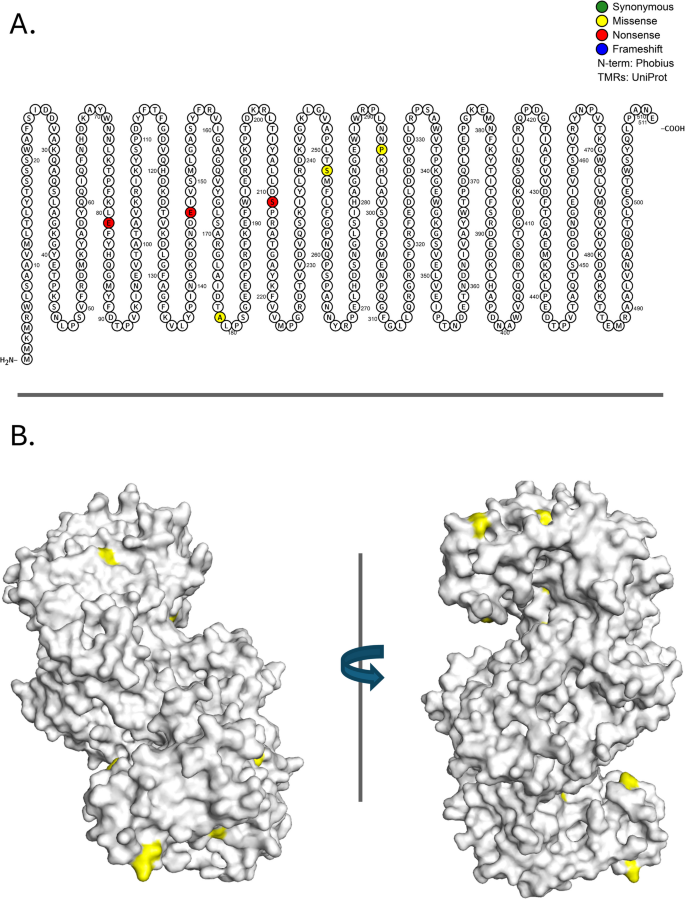
Mutations in proteins required for multifunctional inner membrane protein export: Osmoregulated periplasmic glucans protein, OpgG (N=7, P=1.93E-04). A Location of de novo mutations on the OpgG primary structure. Amino acids that are filled represent the result of a specific mutation that arose during the evolution experiments. The mutation at amino acid position 81 occurred in two chemostats. B Rotated 3-D image of the ancestral genotype with observed missense mutations highlighted in yellow [143, 144]
The adaptive value that enables novel opg alleles repeatedly to spread from single mutant cells to > 2% of a population of 109 cells remains obscure. Blocking the use of glucose as a structural element in the periplasm, as opposed to a source of carbon and energy for growth, could be construed as an energy conservation mechanism. Interestingly, in cells undergoing binary fission under nutrient-rich conditions, OpgH localizes to the nascent septum, where it suppresses assembly of the tubulin-like cell division protein FtsZ [112]. This activity delays cell division and enables cell size to increase. Under slow-growth, nutrient-limiting conditions, there may be a premium for abolishing or diminishing this interaction so as promote cell division among smaller cells.
Lipopolysaccharide (LPS) assembly and transportGenes whose products act in LPS assembly and transport are repeatedly mutated when glucose is limiting
Lipopolysaccharide is an essential component of the E. coli outer membrane, contributing to its structural integrity and providing a protective permeability barrier against a variety of stress factors, including antibiotics and detergents [114]. LPS itself has a tripartite structure, consisting of lipid A (the hydrophobic moiety that anchors LPS to the outer membrane), a core oligosaccharide, and an O-antigen made of repeating oligosaccharide units [114]. The LPS transport system consists of seven proteins (LptA, LptB, LptC, LptD, LptE, LptF, and LptG) that act in concert to extract LPS from the inner membrane then to transport it across the periplasmic space to the outer membrane, where it forms a layer ([114, 115]; Figs. 1C and 10). In our evolution experiments, three of these genes (lptC, lptD, which has significantly clustered mutations, and lptG) are mutated more frequently than expected by chance in both population and clonal sequencing data (Table 1, Additional File 1: Figures S3, S4, S5 and [25]), as is lapB/yciM, which encodes the LPS assembly protein and also has significantly clustered mutations (Additional File 1: Figure S6).
Fig. 10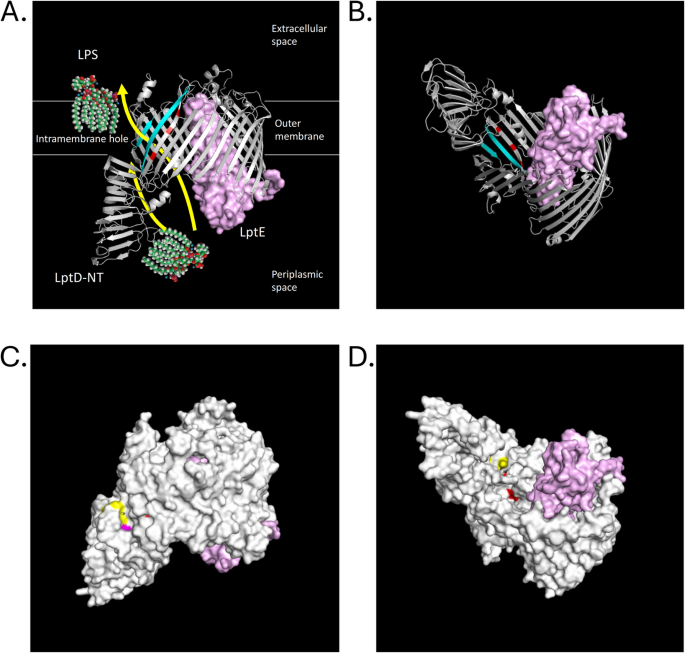
LPS transport is a target of selection under glucose limitation (A) Ribbon view of LPS transport system, and how it extracts LPS from the inner membrane and transport it across the periplasmic space to the outer membrane (also see Figure 1C). B View of A, but from underneath. C A lateral view of LptD/LptE complex (PDB 4Q35 from Shigella flexneri). Hydrophobic residues forming a hydrophobic intra-membrane hole between N-terminus and C-terminus used for lipid A passage are colored in magenta (Trp180, a residue mutated in our evolution to Leu) and yellow (the rest of the residues: Phe203, Phe211, Phe218, Phe228, Leu760 and Leu763, residues in which mutations were not observed). D A view from underneath the LptD barrel (with LptE bound) into a passageway for core oligosaccharide and antigen A portion of LPS. Arg729, Glu733 and Leu736 mutated in our evolution are colored in red and located on the beta26 strand of the luminal gate. An animated 3D image showing de novo mutations in the LptD/LptE complex can be found in Additional File 3: Figure S13. Other LPS transport proteins that are mutated more frequently than can be explained by chance include LptC (N=4, P=1.02E-03), LptD (N=10, P=1.61E-05), LptG (N=7, P=2.24E-05), and LapB (N=7, P=3.64E-05) (see Additional File 1: Figures S3, S4, S5 and S6, respectively)
LptD and LptE form a complex responsible for the final step of transporting LPS to the outer membrane. Assembly protein LptD consists of two domains: the C-terminal half of the protein forms a β-barrel that spans the outer membrane and envelops the LPS assembly protein LptE, while the N-terminal domain is a part of the periplasmic bridge to the inner membrane [116,117,118]. The lipid A part of LPS is transported through the intra-membrane hole formed between the N-terminal and C-terminal portions of LptD, while the O-antigen and the core oligosaccharide are passed between β-strands 1 and 26, opening up the barrel LptD’s C-terminal domain [119]. Both LptE and LptD are considered essential [120].
Crystal structures of the LptD/E complex have been resolved for E. coli (unpublished, reported as PDB 4RHB) as well as those for other Gram-negative bacteria [121]. We mapped de novo mutations in lptD onto structures PDB (Protein Data Bank) 4RHB (LptD C-terminus/LptE complex from E. coli) and PDB 4Q35 (LptD/LptE from Shigella flexneri [121]). Multiple nonsense mutations were located at the extracellular end of the barrel: a single nonsense mutation at Glu676, three independent nonsense mutations at Glu587, and two independent nonsense mutations at Glu618, the last of which is located adjacent to the periplasmic entrance to the barrel, near where LptE is inserted (see Fig. 10C, D as well as an animated 3-D image of the same structure in Additional File 3: Figure S13). It is important to keep in mind that all de novo nonsense mutations recovered in these evolutions may be partly suppressed by a tRNA suppressor mutation in the ancestral strain.
Missense mutations at Gln653 (mutated to His), Ser660 (mutated to Ile), and Ala687 (mutated to Ser) are all located on the same side of the barrel, whereas residues Arg729 (mutated to Leu), Glu733*, and Leu736 (mutated to Met) are accessible from the inside of the barrel’s lumen and are part of β-strand 26, close to where it unzips from β-strand 1. Trp180 (mutated to Leu) has been implicated as providing a hydrophobic intra-membrane exit from the lumen, formed by the N-terminus of LptD, which is used to transport O-antigen and Lipid A (colored in yellow in Fig. 10, Additional File3: Figure S13). Trp180Gln is reported to be lethal in E. coli [119].
LptE stabilizes LptD at the membrane [118] and is required for proper LptD assembly at the membrane [122, 123]. While the observed number of mutations in LptE did not exceed the number expected by chance, both mutated residues in the mature protein (Ser88, Ser125) are located on the protein’s surface (Fig. 10C, D; Additional File 3: Figure S13). Moreover, they cluster with residues in LptE previously implicated in its direct interaction with the lumen of the LptD barrel (Thr86, Phe90, Phe123, Arg124, Met142, Arg150; [117]; Fig. 10). We speculate that decreased LPS biosynthesis may serve as an energy conservation measure under chronic nutrient limitation or that it may play a role in alleviating membrane integrity stress resulting from LamB overproduction [78, 97]. In these respects, it is noteworthy that missense (R165L) and nonsense (E164*) mutations in lptG were fixed in the predominant lineage isolated from an independent E. coli evolution experiment carried out under similar conditions [32].
Proteins required to construct cell surface appendagesMutations in genes required for flagellar synthesis and activity
Twenty-four mutations were observed that are likely to impact flagellar synthesis and activity: 17 occurred in the fliFGHIJK operon, of which 11 were nonsense mutations, while 7 occurred in fliMNOPQR operon, of which 2 were nonsense [25]. Of these twelve loci, three were mutated more frequently than expected by chance: fliG, fliH, and fliP, with fliG showing significantly clustered mutations (Table 1, Fig. 1D, and Additional File 1: Figures S7, S8, S9). fliP and fliH encode components of the flagellar export apparatus: FliP being a cytoplasmic ATPase (Adenosine triphosphatase), FliHbeingone of six integral membrane components. All five fliH alleles were transversions, 4 of which resulted in nonsense mutations (Glu37*, Glu62*, Glu104*, Cys220*); none of these alleles ever exceeded 13% frequency in our experimental populations [25].
FliG, FliM, and FliN form the C-ring of the flagellar motor switch ([124]; Fig. 1D). FliG consists of 331 amino acid residues organized in at least two discrete domains: one at the carboxy terminus, another in the middle of the protein. The C-terminal domain of ~ 100 residues is essential for flagellar rotation, but dispensable for flagellar assembly [125]. The flagellar rotor itself consists of ~ 25 FliG molecules that interact with one another and with FliM, which in turn interacts with the FliN C-ring protein. It is in the FliG middle domain where FliG:FliG and FliG:FliM interactions appear to occur [125]. In our experiments all five de novo fliG mutations were GT transversions, four of which resulted in nonsense mutations: Glu19*, Glu19*, Glu174*, Glu177*, and one of which resulted in a missense mutation from a non-polar to a polar residue, Ala173Ser (Additional File 1: Figure S7). Three of these mutations were found to cluster between amino acids 172 and 177 by the Cluster Explorer and NMC algorithms (Table 1). The phenotype and severity of these C-terminal and middle domain nonsense mutations would depend on the efficiency with which the ancestral tRNA glnX suppressor enables full-length FliG protein to be translated. Finally, it should be noted that in addition to mutations in the fliMNOPQRoperon de novo mutations also occurred at greater than expected frequencies in the gene encoding the flagellar biosynthesis protein, FlhB as well as in the gene encoding the flagellar assembly protein, FlgJ (Additional File 1: Figures S10 and S11; Additional File 2: Table S1 and [25]).
In E. coli motility is linked to growth rate through the flagellar master regulator FlhD4C2, and cells grown in glucose-limited chemostats at slow dilution rate (µ = 0.12 h−1) exhibit produce fewer flagella than cells grown at high dilution rate (µ = 0.6 h−1) [126]. This observation suggests a trade-off between resource investment in motility versus growth in a nutrient-poor environment, especially when that environment is well-mixed, as is the case in a chemostat [127]. Indeed, flagellar biosynthesis may draw upon as much as 2% of an E. coli cell’s biosynthetic capacity and 0.1% of its total energy expenditure [128]. Diminished expression of E. coli flagellar operons has also been seen in long-term evolution experiments carried out via serial dilution [129]. And, in previous chemostat experiments originating from the same ancestor used here, multiple mutations in fliF, fliH, flI, and fliM were observed, with three (E59D, A62S, E178*) occurring in the dominant lineage at the motor switching and energizing component, fliM [32]. We speculate that recurrent mutations in flagellar operons may be due to two related factors: reduced investment in the motility apparatus among cells growing slowly (D = 0.2 h−1) in a well-mixed, nutrient-poor environment and followed by selection on the motility apparatus because it is weakly expressed and nonessential under these conditions.
Mutations required for type 1 fimbriae secretion are frequently selected, inducing biofilm formation pathways
While biofilm formation was not an intended outcome of selection under continuous glucose limitation, it appears to be an adaptive strategy that enables low-frequency clones to persist. We observed biofilms in each of our replicate evolutions, and our genetic data reflect that observation. Type 1 fimbriae are required to maintain biofilm structures in E. coli [130], and expression of the fim operon (fimAICDFGH) encoding the structural components of type 1 pili is regulated by an invertible switch, fimS ([131, 132]; Fig. 11A). fimS controls type 1 pilin phase variation in E. coli and consists of two recombinase genes, fimB and fimE, that lie immediately upstream of fimS. FimE protein is responsible for switching fimS from “Phase ON” to “Phase OFF” while FimB functions as a bi-directional recombinase [133,134,135]. In our experiments, fimS remained in the ancestral “off” configuration for the great majority of sequenced clones. However, we did observe a small minority of clones that had inverted fimS in the Phase ON orientation (see Methods), which is expected to result in transcription of the fim operon. We suggest that selection of the chromosomal inversion activating fimS primes “Phase ON” lineages for selection of additional fim operon mutations (e.g., fimH, Additional File 1: Figure S12 and discussion below).
Fig. 11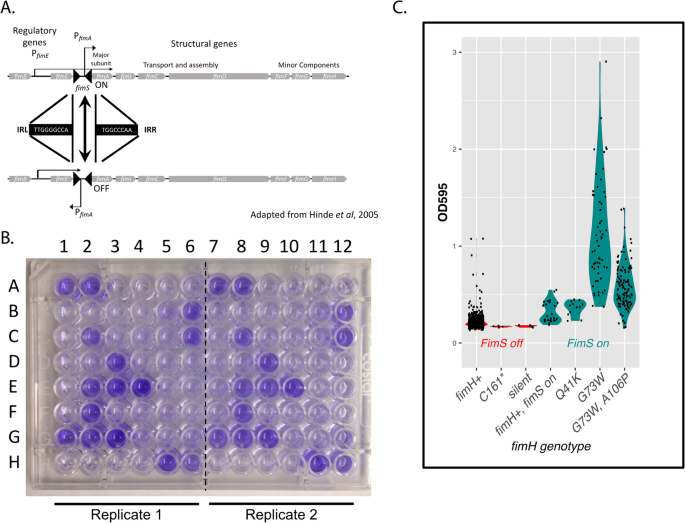
Genes required for biofilm formation are mutated in glucose-limited E. coli populations. A The Fim switch and transcriptional regulation of the Fim operon. The region bracketed by filled triangles represents the invertible fim switch (fimS). When the switch is in the top configuration, the fim operon is transcribed (ON). When the switch is in the bottom configuration (inverted), the fim operon is not transcribed (OFF). B Microtiter plate with clones A1-6 through H1-6 from chemostat 2 (columns 1-6 are technical replicates of columns 7-12) grown, washed and stained with Crystal Violet dye (see Methods). The propensity to form biofilms positively correlates with the intensity of color. C Biofilm formation in clones with activated form of fim operon switch (fimS ON, in green) and deactivated (fimS OFF, in red), and various fimH genotypes. The propensity to form biofilms was evaluated in quadruplicate by measuring optical density at 595nm. Clones with identical genotypes that arose independently in different chemostats are plotted separately. Evolved clones with the Fim operon turned ON differ significantly from wild-type clones whose Fim operon is turned OFF (p-value < 2.5E-09)
Fimbriated cells normally grow more slowly than those that do not produce fimbriae, especially at lower temperatures (our experiments were carried out at 30 °C not 37 °C) [136]. However, in a chemostat the growth rate cost of making fimbriae might be offset by the benefit of increased adherence to vessel walls, which would extend cells’ residence time and spatially segregate them from the larger—and more rapidly dividing—planktonic population. While simply turning on the fim operon likely provides some selective advantage, most sequenced clones with the fimS switch turned on (43 out of 55) possessed additional mutations in the fimH gene encoded within the fim operon (Fig. 1D and below). Mutant fimH clones tested strongly positive for biofilm formation in a microtiter plate assay, while wild type fimH clones did not (Figs. 11 and 12). In 12 of 55 fimS “Phase ON” clones that lack additional fim operon mutations we sometimes observed other biofilm-relevant mutations. For example, three mutations (Pro30Thr, Ala102Glu, and Ala157Asp) were observed in matA, which encodes a transcription factor that exerts a dual regulatory function on the choice of planktonic vs. sessile lifestyle [137]. There is also a matA promoter mutation in 4 clones from chemostat 3 (G4, E1, D8, C6). How (or whether) these mutations affect matA expression is unknown (Additional File 2: TableS1).
Fig. 12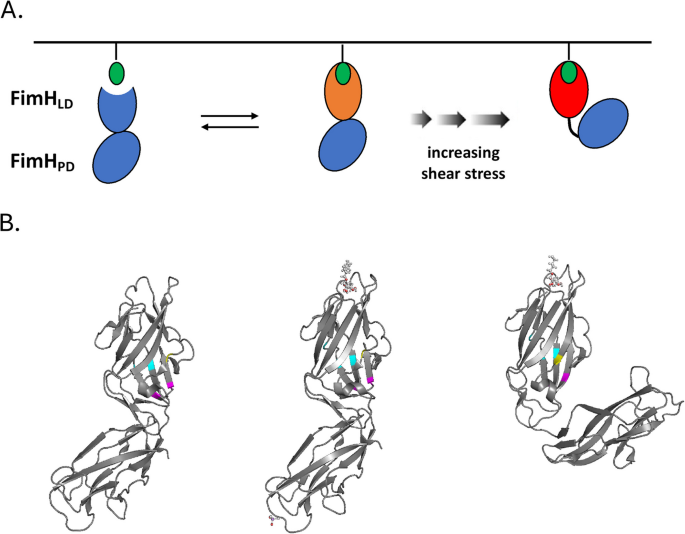
E. coli FimH undergoes allosteric changes in response to lectin binding and shear stress that affect its propensity to adhere to the uroepithelium and promote biofilm formation. A Full length FimHFL consists of an N-terminal lectin domain (FimHLD) connected to a C-terminal pilin domain (FimHPD), which undergo conformational changes upon binding to mannosylated uroplakin 1a (UP1a), whose sugar moiety projects from the luminal side of the urothelium (green). FimHFL undergoes additional changes in response to shear stress (adapted from [136]). B FimH structure bound to n-heptyl-α-d-mannopyranoside in the low-affinity state (PDB 4XOE) (left), in the high-affinity state (middle), and in the presence of shear force (PDB 4XOB) (right). Asn33 and Gly73 are represented by magenta, Asp37, Gln41 and Ala106 by cyan, Ser62 by yellow. Mutants at each of these positions were recovered from our evolution experiments as well as from screens of uropathogenic strains. (Also see Additional File 4: Figure S14, an animated 3-D image of FimH showing the location of de novo mutations recovered in our experiments.)
FimH mutations arising in E. coli under glucose limitation in the lab recapitulate FimH mutations seen in pathogenic E. coli isolated in the clinic
FimH encodes a type 1 fimbrial adhesin that binds D-mannose [138] (Figs. 1D and 11). The majority of fimS “ON” clones contained additional mutations in FimH, with three residues in FimH (Asp37, Gln41, and Gly73) being recurrent, independent targets of mutation, suggesting that these mutations are adaptive. (Note: The amino acid numbering used here reflects cleavage of the signal peptide [139], and therefore does not match the numbering in Additional File 2:Table S1). We also observed additional single mutations affecting two other residues (Asn33 and Ala106). Asn33His and Gly73Glu have both been previously observed as naturally occurring variants in uropathogenic strains (CI#7 and CI#4 respectively); both bind yeast mannan and human fibronectin [140]. FimH mutants containing either Asn33His or Gly73Glu exhibit higher affinity to monomeric mannose relative to an otherwise isogenic strain lacking those mutations [141]. Importantly, higher affinity to monomeric mannose, compared to tri-mannose structures, differentiates multiple uropathogenic strains from bowel isolates originating in healthy individuals, and has been shown to provide a selective advantage for urinary tract colonization in mice [141]. Similarly, Gly73Glu mutants were recovered in a screen for clones from a fimH mutant library that more readily agglutinate yeast and exhibit higher affinity for human fibronectin [142]. Last, Gly73 and Ala106 in FimH have been described as evolutionary hot spots in E. coli strains isolated from patients with Crohn’s disease. Gly73Arg, Gly73Glu, Gly73Ala, Gly73Trp (the exact change that we observed), and Ala106Trp substitutions have been seen among Crohn’s isolates able to bind human T84 intestinal epithelial cells and cause inflammatory bowel disorder [143]. Together, these observations show that our chemostat evolutions select for clinically relevant mutations in fimH related to biofilm formation.
To better understand how these mutations might result in a biofilm phenotype, we modeled how they affect FimH structure. The FimH protein consists of lectin and pilin domains that adopt different conformations, depending on the presence/magnitude of shear stress induced by flow of fluids [144, 145]. When the lectin domain is bound to mannose, the lectin and pilin domains are connected more rigidly in the absence of flow. However, in the presence of flow the connection between the two domains becomes more flexible, increasing ligand affinity [144]. We examined the locations of the observed mutated residues in the structures of both conformations (see Fig. 12, Additional File 1: Figure S12 plus animated 3-D image of FimH structure Additional File4: Figure S14). Asn33 and Gly73 exhibit different accessibility between the two conformational states. In the relaxed, high-affinity conformation (with shear stress) they are exposed, while in the rigid, low-affinity conformation they are almost entirely buried (in magenta, Fig. 12B). This is similar to another residue (Ser62) located in the same vicinity, which has also been identified as polymorphic in multiple pathogenic E. coli strains (uropathogenic strain NU14 and neonatal meningitis isolates RS218 and IHE3034; [146]; Fig. 12B in yellow). While accessibility of the other residues that we observed as mutated (Asp37, Gln41, Ala106) does not vary between conformations, they do cluster together within the structure (Fig. 12B in cyan), suggesting they may impact activity of the FimH adhesin via a similar mechanism.