
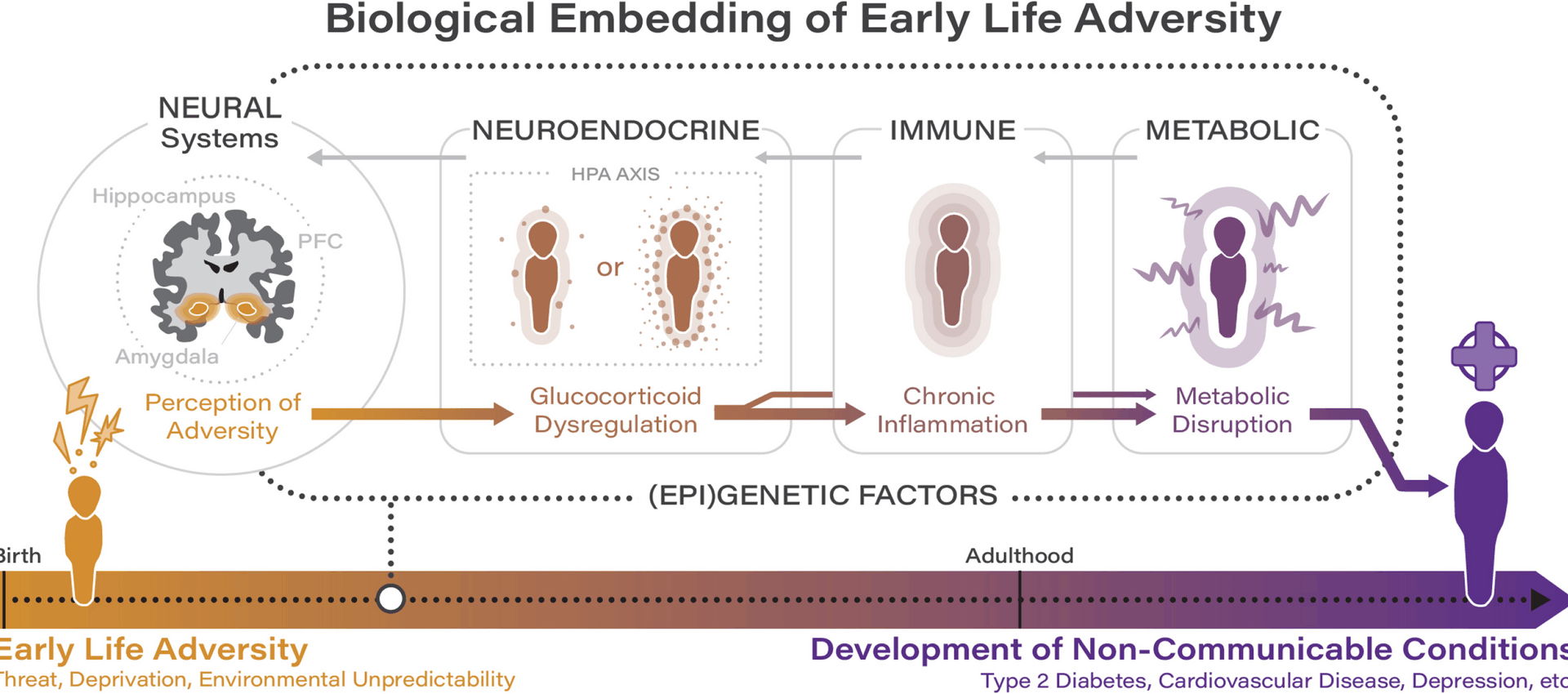
Early life adversity has a profound and life-long impact on health and development, initiating a cascade of dysregulations involving epigenetic and neural mechanisms across multiple interconnected biological systems. In this section, we consolidate recent evidence to outline a developmental cascade model illustrating how disruptions in one system—precipitated by early life adversity—can have a cumulative impact on health trajectories (see Fig. 1). This model provides a comprehensive framework for understanding how early life adversity becomes biologically embedded and perpetuates risk for non-communicable conditions across the lifespan. While numerous physiological systems are implicated in the embedding of adversity, we focus on the role of the neuroendocrine, immune, and metabolic systems given their central role in stress physiology and their consistent identification in the literature as key pathways through which early life adversity impacts long-term health.
Fig. 1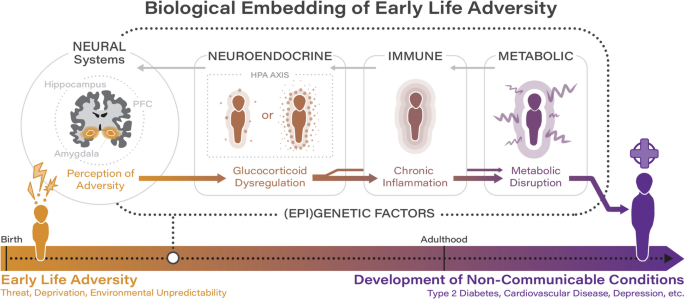
Recent evidence regarding the biological embedding of adversity implicates dysregulations across the neuroendocrine, immune, and metabolic systems, which bidirectionally influence and are influenced by neural and (epi)genetic factors
As argued by Hertzman [10], a core neurobiological system involved in the biological embedding of stress is the hypothalamic–pituitary–adrenal (HPA) axis. The HPA axis is a primary stress response system that is part of the broader neuroendocrine system whose activation results in the secretion of cortisol in humans [16]. Indeed, of all systems impacted by early life adversity, the HPA axis is one of the most well-studied. Early life adversity is processed through central neurobiological circuits, where experiences of adversity are encoded and initiate responses that activate the HPA axis through inputs involving the prefrontal cortex, amygdala, and hippocampus [17]. Although it is possible that the direction of dysregulation may differ depending on the type and timing of adversity [18], early life adversity—and particularly adversity characterized by threat and harshness, such as neighborhood violence (distal ecological exposure) and physical abuse (proximal direct exposure)—has broadly been associated with cortisol hypersecretion during childhood and adolescence [19,20,21]. From an evolutionary perspective and in line with life history theory, heightened vigilance and stress system responsivity in a dangerous and unpredictable environment is likely to be highly adaptive [22, 23]. However, when chronic, the hypersecretion of cortisol has been shown to disrupt the development of brain regions responsible for stress regulation and emotional processing including the prefrontal cortex, amygdala, and hippocampus, given the high degree of plasticity inherent in the developing brain and the high density of glucocorticoid receptors in these regions [24, 25]. Indeed, recent work by Meaney and colleagues integrated transcriptomic data from model systems with human genomics to position hippocampal glucocorticoid signaling as a mediator of the association between early life adversity and psychopathology [26]. By using transcriptomic data from model systems to identify gene-network signatures of susceptibility to early life adversity, this work highlights the value of incorporating genetic and epigenetic methodologies into the study of early life adversity and non-communicable health outcomes. Such approaches are particularly exciting in the context of neuroendocrine dysregulations, as they have the potential to help disentangle the specific role of cortisol in the development of non-communicable conditions given mixed findings in this area. While many studies do provide evidence of HPA axis hyperactivity in response to early adversity, others report blunted cortisol profiles, particularly in older adolescents and adults, which raises important questions about developmental shifts and the possible emergence of hypocortisolism as an adaptation to prolonged stress [27, 28]. These mixed findings likely reflect multiple, non-exclusive factors such as the developmental timing of assessments; chronicity and type of adversity experienced; methodological variation in sampling and assay procedures; and genetic and epigenetic differences in stress-regulatory pathways. Future genetic and epigenetic work could disentangle these patterns by examining whether variants or epigenetic profiles (e.g., methylation states) of HPA-axis genes (e.g., FKBP5) and polygenic stress-sensitivity scores moderate the direction and magnitude of HPA axis activity across developmental stages. Thus, (epi)genetic stratification—when conducted with careful attention to factors including developmental timing, chronicity/type of adversity, and methodological variation—has the potential to illuminate why some individuals demonstrate cortisol hyperactivity while others demonstrate hyporeactivity following exposure to ELA. This enhanced understanding could advance our ability to inform personalized intervention and prevention strategies targeting specific biological pathways among individuals affected by ELA.
In well-regulated systems, the presence of glucocorticoids (cortisol in humans) has an anti-inflammatory effect by downregulating the expression of inflammatory proteins through the inhibition of pro-inflammatory gene transcription [29]. Glucocorticoid-based inhibition of immune responsivity is recognized as the most fundamental physiological mechanism for protection against diseases that involve excessive inflammation. However, a recent systematic review across species suggests that chronic HPA-axis hyperactivity, as is seen following ELA, can result in a downregulation of intracellular glucocorticoid receptor sensitivity to glucocorticoids, which leads to an upregulationin pro-inflammatory gene transcription and ultimately elevated inflammatory activity [30]. In other words, cortisol can lose its typical anti-inflammatory effects, resulting in increased inflammation through stress-related dysregulation in cellular glucocorticoid sensitivity (i.e., glucocorticoid resistance). This is illustrated by findings demonstrating that children exposed to high levels of prenatal maternal stress and to chronic stressors during adolescence exhibit both elevated cortisol reactivity and increases in pro-inflammatory cytokines [31,32,33]. However, it is important to note that some studies have reported inconsistent or null associations between early adversity and inflammation, or have found that associations are moderated by subsequent environmental exposures and/or health behaviors, which underscores the complex and multifactorial nature of these pathways as well as the methodological variability present in the literature [34, 35].
From a life history perspective, glucocorticoid resistance can be seen as an adaptive response to early adversity that is consistent with a “fast” life history strategy: in a dangerous or unpredictable environment, it becomes adaptive from an evolutionary standpoint to shift energy away from long-term maintenance and toward immediate survival [23, 36]. Heightened inflammation, even if harmful long-term, provides short-term benefits by enhancing an organism’s ability to heal wounds and fight infections that are more common in dangerous and unpredictable environments. Thus, glucocorticoid resistance represents a biological trade-off: in response to persistent threats, the immune system favors increased vigilance at the cost of chronically elevated inflammation. In the context of bench-to-bedside translation, it will be important in future work to determine whether it is possible to identify a physiological “tipping point” in the transition towards a glucocorticoid-related pro-inflammatory phenotype.
This work contributes to an increasingly sophisticated understanding of the link between early life adversity and immune system dysfunction. The human immune system comprises both innate and adaptive branches. Together, early adversity impacts both the fast-acting innate and the long-lasting adaptive branches of the immune system, resulting in heightened and sustained inflammatory activity that catalyzes downstream risk for non-communicable health outcomes. For instance, recent work by Rasmussen and colleagues has linked exposure to early life adversity to elevated levels of suPAR, a novel biomarker associated with inflammation and immune activation whose initial release is driven by the innate immune system while sustained or amplified production may instead be driven by adaptive immunity [37, 38]. In turn, heightened levels of suPAR indicate a heightened inflammatory state and predict morbidity and mortality associated with multiple non-communicable conditions including cardiovascular disease, type 2 diabetes mellitus, and depression [39, 40]. Further, extending prior work demonstrating cross-sectional associations between early life adversity and inflammatory markers, Renna et al. recently demonstrated that individuals with a history of early life adversity demonstrated a steeper increase in an inflammatory composite across a 2-year period during adulthood [41]. Alongside dysregulations in glucocorticoid sensitivity, adversity-related differences in DNA methylation in stress-related genes and those involved in the regulation of inflammation likely contribute to increases in inflammation [42]. Through these mechanisms, immune dysfunction compounds HPA axis dysfunction and primes inflammatory feedback loops that converge on metabolic pathways.
Alongside the neuroendocrine and immune systems, the metabolic system plays a key role in connecting early life adversity with risk for non-communicable conditions. This is supported by a recent meta-analysis across 13 international cohorts (N= 217,929) that documented progressively higher odds of cardiometabolic disease among individuals with a history of childhood maltreatment [43]. Current evidence indicates that impaired glucose regulation and insulin sensitivity (critical indicators of metabolic health underlying risk for cardiometabolic disease) following adversity are likely related to a complex interaction of neuroendocrine and inflammatory processes. Indeed, both chronically high glucocorticoid levels and chronic low-grade inflammation have been associated with metabolic disturbances such as dyslipidaemia, hyperglycaemia, and insulin resistance, in part by increasing circulating free fatty acids and through the induction of lipid accumulation and impairments in adipocyte differentiation [44, 45]. There is also recent evidence to suggest that the association between early life adversity and markers of metabolic dysfunction is mediated by epigenetic mechanisms. For instance, recent work by Tobi and colleagues indicates that DNA methylation mediates the impact of prenatal adversity on metabolic outcomes including elevated body mass index (BMI) and serum triglycerides [46]. This work highlights epigenetics as an important interface through which genetics and early life adversity converge to influence risk for non-communicable conditions, such as cardiometabolic disease, across the lifespan.
In sum, recent work indicates that exposure to many forms of early adversity can result in chronic HPA axis dysregulation, leading to a reduced physiological sensitivity to cortisol. Epigenetic modifications, including DNA methylation changes in stress-related genes, further contribute to alterations in glucocorticoid signaling and immune regulation. Together with epigenetic changes, this glucocorticoid resistance impairs the body’s ability to regulate immune function. In turn, chronic inflammation resulting from heightened pro-inflammatory activity worsens metabolic disruption, exacerbating glucose dysregulation and promoting insulin resistance. In feedback loops with the developing brain, stress regulatory systems become increasingly impacted, exacerbating physiological and psychological vulnerabilities that can accumulate and manifest as non-communicable conditions. Thus, rather than isolated systems-level impacts, recent work supports a cumulative progression of dysregulation driven by early adversity-driven programming. However, as highlighted previously, the pathway from early life adversity to illness is not uniform, and null or contrary results in the literature—likely attributable to variation in assessment approaches, sample heterogeneity, and unconsidered moderating factors—underscore the need for studies that employ rigorous longitudinal and genetically informed designs to clarify the boundaries and mechanisms of effects. Indeed, the pathway to illness is not linear and involves a complex interplay of systems-level factors which interact with behavioral pathways to non-communicable health outcomes (such as sedentary lifestyles, poor diet, medical nonadherence, sleep disturbances, smoking, and substance use, including excess alcohol consumption), which contribute to and are impacted by these cascading pathways, further exacerbating risk for disease.
Opportunities for a maturing field
These findings, which largely focus on the integration of multiple biological systems, underscore the potential for early life adversity to impact biological systems in a dynamic and multi-system manner and mark a shift away from single biomarker approaches towards integrated models of lifespan biological embedding. Despite the enormous promise of this work for continued scientific progress, the degree to which findings inform clinical practice remains limited. We contend that four key future directions must be emphasized in future work to both ensure continued scientific progress and maximize the clinical impact of the field (see Fig. 2).
Fig. 2
Summary of key future directions for the field concerned with the role of early life adversity in contributing to the development of non-communicable health outcomes
First, research in the field concerned with the impact of early life adversity on risk for non-communicable health outcomes has traditionally been siloed, focused on either physical or mental manifestations of illness, and is often disease specific. Cross-disciplinary research that reaches across traditional academic boundaries is crucial to support the identification of common biological mechanisms or moderators that will arguably hold the most promise for the development of potent biological intervention strategies. Indeed, high rates of comorbidity between psychiatric and physical manifestations of illness (such as depression and cardiometabolic disease) are often highlighted in the literature; while these rates speak to the likely existence of common underlying mechanisms or risk factors, very few studies explicitly explore this possibility. One particularly exciting opportunity for collaboration involves primary care clinics (i.e., medical clinics that offer a variety of health services, including preventive care, routine checkups, and treatment for common illnesses), which are uniquely positioned at the intersection of physical and mental healthcare. Primary care clinics encounter youth in subclinical ranges or early in the course of illness, which allows for the opportunity to screen for early life adversity during sensitive developmental windows when neurobiological systems are most plastic and responsive to intervention. Clinics can then implement developmentally informed interventions aimed at buffering youth against the neurobiological impacts of adversity. Indeed, in line with recent work, we argue that information related to experiences of early life adversity should be considered as important medical information that should be routinely collected in care settings [47]. A current initiative adopted in California, ACEs Aware, has demonstrated that statewide screening initiatives situated within healthcare settings are highly feasible and scalable [48, 49]. Further, primary care clinics not only support efficient biomarker assessment through routine screening and point-of-care testing, but also facilitate access to electronic health records (EHRs), which enables researchers to leverage existing longitudinal data to track the progression of underlying processes linked to early life adversity and non-communicable health outcomes across childhood and adolescence, when physiological systems are undergoing rapid change. Further, primary care clinics often serve a broad population and thus provide important access to youth and families with diverse socioeconomic, racial, and ethnic backgrounds. This not only increases the generalizability of results to the broader population but also means that research taking place in this setting can identify how adversity interacts with developmental timing across varied populations to identify equity focused solutions to reduce disparities in physical and mental health outcomes for populations that are disproportionality impacted by early life adversity. Finally, findings from collaborations involving primary care settings can be directly translated into clinical workflows, personalizing interventions based on transdiagnostic insights, and enhancing screening for experiences of early life adversity.
Second, it is crucial that the field continues to move beyond the single biomarker approach (i.e., to integrate multiple biomarkers) to model dynamic cross-system integrations linking early life adversity to risk for non-communicable conditions. Indeed, as evidenced by recent work, the biological pathways linking early adversity to pernicious outcomes are complex, interdependent, and involve multiple systems [23, 33, 34]. Single biomarker approaches cannot capture this complexity, nor can they model the temporal dynamics of multi-system dysregulations. Multi-biomarker approaches are not only possible but increasingly feasible given advances in multi-omics data integration involving advanced bioinformatics tools and machine learning, as well as the increased availability of minimally invasive options for the assessment of biomarkers [50, 51]. For instance, non-invasive collection methods such as saliva, sweat, and hair sampling have provided accessible alternatives to serum-based biomarkers, while dried blood spots allow for the remote collection of blood samples, enabling assessments of neuroendocrine, inflammatory, and metabolic indicators. Wearable biomarker technologies offer further opportunities for the assessment of physiological markers that can complement traditional biomarker measures derived from fluid and tissue samples. Systems-level insights arising from multi-biomarker approaches support the characterization of biomarker signatures associated with exposure to early adversity that are predictive of risk for non-communicable conditions and progression, even prior to the emergence of clinical symptoms, which facilitates prevention, early detection, and intervention. Recent methodological advances have also addressed many prior challenges in biomarker assessment, resulting in the development of best-practice guidelines for sample collection, storage, and processing for many commonly used biomarkers such as cortisol, proinflammatory cytokines, and telomeres, among others [52,53,54]. Attention to study design is also essential for improving reproducibility and translational potential. Careful selection of sample type based on research question and biomarker stability, clear reporting of collection context and timing (e.g., circadian considerations for cortisol), protocols to minimize degradation and contamination, and use of duplicate samples or longitudinal sampling designs can mitigate assessment error and improve interpretability. International research networks, such as the Telomere Research Network, are setting the standard by providing harmonized assessment protocols and reporting guidelines which have enormous promise for enhancing the scientific rigor and reproducibility of the field.
Indeed, innovations over the past decade have made it possible to characterize disease-related biological processes prior to the onset of disease [13,14,15], which affords the opportunity for more effective preventative healthcare strategies. For instance, it is possible to quantify processes linked to age-related disease among youth and young adults for whom there is the possibility of delaying or even reversing the “march” toward disease onset [55,56,57,58]. Further, by integrating data from multiple biological layers (or “omes,” such as epigenomics, transcriptomics, proteomics, and metabolomics), multi-omics approaches provide a comprehensive view of the molecular changes occurring within an individual and can identify molecular signatures unique to specific developmental periods following adversity exposure. This integration enables the identification of early disease processes. For instance, recent work by Wainberg and colleagues integrated genomic, proteomic, and metabolomic data to uncover detectable disease-risk signatures among a relatively healthy sample [59]. Such an approach could be applied to samples of individuals with a history of early life adversity by treating adversity-specific variables as key modifiers in analyses or by using adversity exposure data to inform interactions between genetic predisposition (e.g., polygenic risk scores, or the weighted sum of risk alleles carried by an individual) [60] and adversity-related molecular signatures. A recent review by Layfield and colleagues considers contemporary work demonstrating the potential of multi-omic approaches for improving our understanding of the distinct mechanisms contributing to risk versus resilience in response to early adversity [61]. However, as biomarker discovery advances, the field must also address ongoing challenges in causal inference, which is critical for translating biological insights into effective, mechanistically informed interventions. Correlational findings alone are insufficient to support mechanistic claims, particularly in the context of epigenetics or other ‘omics-based fields in which the directionality of effects often remain ambiguous. Further, future work must account for gene-environment correlations (rGEs) which can inflate associations between adversity and later outcomes. As an example of passive rGEs, a parent with depression could pass on a genetic risk for depression and their depression contribute to an adverse environment for the child. Thus, though early adversity may be associated with depression, a causal path cannot be assumed given that some of this association could be attributed to shared genetic risk. Further, recent meta-analytic work estimates that the association between childhood maltreatment and mental health outcomes in quasi-experimental studies is substantially (45%) smaller in adjusted models compared to unadjusted models [62]. This provides further support to the notion that a significant proportion of the association between ELA and non-communicable health outcomes is confounded by preexisting (genetic) risk. This highlights the importance of genetically informed designs, such as twin and adoption studies, sibling control analyses, and intergenerational Mendelian randomization, which can parse the contributions of genetics, shared versus distinct environments, and individual-specific environmental factors in associations between early adversity and non-communicable conditions such as major depressive disorder and T2DM. Further, integrating polygenic risk scores into multi-omics studies can elucidate how genetic predispositions interact with early environmental exposures to shape biological trajectories. Given that each method has distinct strengths and limitations, prospective triangulation designs that integrate multiple genetically informed approaches are particularly well suited to disentangling complex gene-environment interplay and strengthening causal interference, with the aim of advancing our understanding of how early life adversity impacts risk for non-communicable conditions [63].
Recent empirical and theoretical work highlights the importance of moving beyond cumulative and adversity-specific models to instead conceptualize early life adversity in ways that support the identification of underlying mechanisms through which dimensions of adverse experiences influence distinct features of development (see Fig. 3). Early work in the field conceptualized early life adversity according to a cumulative model, using a summed score based on total exposure to adverse events endorsed by an individual. While this work catalyzed the field concerned with the long-term implications of early adversity, cumulative models have not provided strong evidence for potential mechanisms linking exposure to adversity early in life with negative outcomes, nor do they consider the severity, timing, or duration of adversity [64]. Moving beyond this approach, research stimulated by specificity models has focused on the impacts of specific forms of adversity such as caregiver psychiatric illness, neglect, physical abuse, sexual abuse, and poverty. However, as reviewed in McLaughlin et al., specificity models do not acknowledge the high rates of co-occurrence among forms of early adversity and fail to recognize that there may exist common underlying mechanisms linking different forms of early adversity with outcomes [65]. In response to these limitations, dimensional models have emerged as promising alternatives to cumulative and specificity approaches [23, 66, 67]. Grounded in the idea that core dimensions of early experience underlie multiple types of adversity, dimensional models focus on aspects of environmental exposures that can be conceptualized along a continuum, such as threat, deprivation, harshness, and unpredictability [68]. For instance, early exposures characterized by threat include witnessing domestic violence and physical abuse, while early exposures characterized by threat include neglect and institutional rearing [69, 70]. By disaggregating adversities, dimensional models highlight unique and shared pathways to illness to clarify how distinct aspects of experiences and exposures become biologically embedded, ultimately allowing for the identification of more precise biomarkers and intervention targets which could lead to more effective prevention and treatment strategies for non-communicable health outcomes rooted in early life adversity. Dimensional models, when combined with genetically sensitive and mediation-based study designs, offer a particularly powerful framework for identifying mechanisms of risk and resilience capable of informing both policy and practice. Regarding the operationalization of early life adversity, there have been many lessons learned over the past several decades with regard to fundamental issues that must be considered when assessing exposure to early adversity. A particularly critical consideration is the use of prospective versus retrospective measures of early life adversity. Two recent systematic reviews and meta-analyses conducted by Baldwin et al. [71, 72] suggest limited agreement between prospective and retrospective measures of childhood maltreatment, finding that these measures largely identify different groups of individuals and that retrospective measures of childhood maltreatment show stronger associations with psychopathology compared to prospective measures. Important considerations including prospective versus retrospective reporting, objective versus subjective assessment approaches, and caregiver versus child reporting, as well as best practices in the assessment of ELA, are discussed in several recent reviews [73, 74]. Collectively, the current literature underscores the importance of a nuanced, multidimensional approach if we are to unravel the complex ways in which early life adversity shapes developmental trajectories. By continuing to refine our conceptualization and measurement of early life adversity, the field is poised to identify unique aspects of experience that influence behavioral and neurobiological development to delineate specific mechanisms through which early adversity contributes to a neurobiological foundation underlying risk for non-communicable health outcomes.
Fig. 3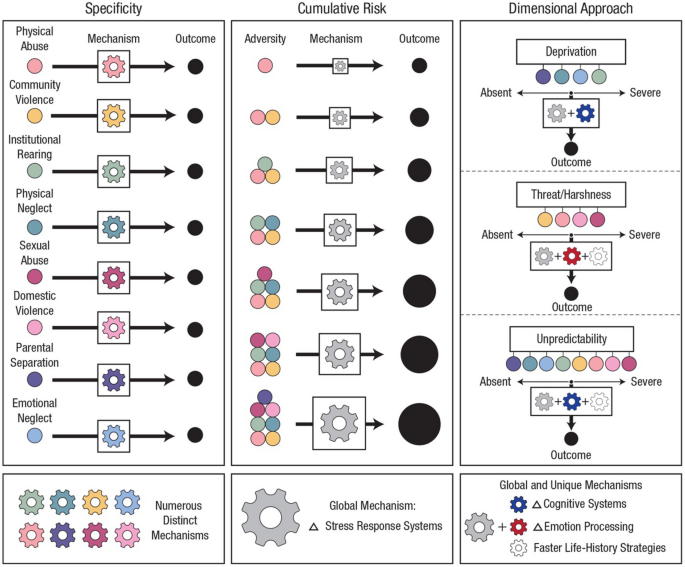
Approaches to conceptualization of early life adversity. Figure built in collaboration with Nessa Bryce. Reproduced, with permission, from [65]