
DNA extraction and sequencing
For long-read sequencing, we began with 3-week-old plants grown in soil that had been transferred to darkness for 24–48 h before harvesting to reduce the starch content. A total of 20–30 g of flash-frozen rosette tissue, pooled from individuals, was ground in liquid nitrogen with a pestle and mortar. Nuclei were isolated as described for accession Ey15-2 (ref. 75), and high-molecular-weight (HMW) DNA purified with Genomic-tips 100G (Qiagen, 10243) following the manufacturer’s instructions. Ten micrograms of HMW DNA were sheared with either Megaruptor 3 (Diagenode, B06010003) or a FINE-JECT 26G× 1″ needle (0.45 × 25 mm; 14-13651) to ca. 75 kb, and used as input for long-read library preparation with the SMRTbell Express Template Preparation Kit 2.0 (Pacific Biosciences, 101-693-800). These libraries were size-selected with the BluePippin system (Sage Science) with a 30 kb cutoff in a 0.75% DF Marker U1 high-pass 30–40 kb vs3 gel cassette (Biozym, BLF7510). Libraries for accessions 9981 (Angit-1; CS76366) and 10002 (TueWal-2; CS76405) were sequenced on a Sequel II system (Pacific Biosciences), and the others on a Sequel I system.
To prepare PCR-free libraries for short-read sequencing, the genomic DNA was fragmented to 250–350 bp using a Covaris S2 Focused Ultrasonicator (Covaris). The libraries were prepared according to the manufacturer’s instructions with either the TruSeq DNA PCR-free kit (Illumina, 20015962) or the NxSeq AmpFREE Low DNA Library kit (Lucigen, 14000-2). In total, libraries for 89 accessions (including the main 27 for which we assembled their genomes) were sequenced in paired-end mode on a HiSeq 3000 system (Illumina).
The ultra-HMW DNA extraction and sample preparation for optical maps were performed as described75,76 at Corteva Agriscience using the Direct Label and Stain technology (Bionano Genomics).
Assembly
The CLR subreads were assembled with Canu v1.71 (ref. 77). Since accessions 9981 and 10002 had been sequenced at higher coverage on a Sequel II instrument, only about 200× genome coverage worth of reads were used for assembly. We performed two rounds of polishing on the resulting contigs of all assemblies—first with the CLR subreads and Arrow v2.3.2 (https://github.com/PacificBiosciences/gcpp), and then with PCR-free short reads and Pilon v1.22 (ref. 78).
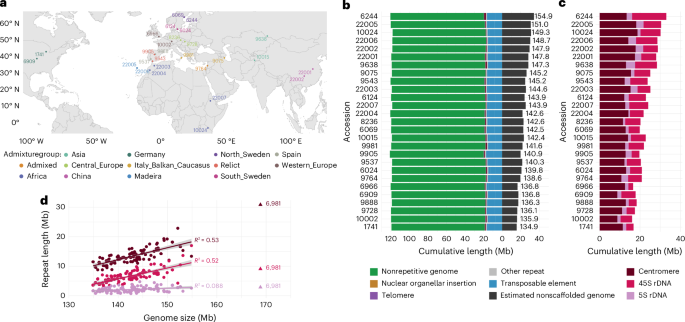
For scaffolding, we generated hybrid scaffolds with optical maps for eight accessions (Supplementary Table 1) using Bionano Access v1.5 and Bionano Solve v3.6. The assembly was performed in pre-assembly mode with parameters nonhaplotype and no-CMPR-cut, without extend-split. Based on what we learned from these hybrid assemblies, we set the parameters for in silico scaffolding of the other genomes. We scaffolded contigs >150 kb with RagTag v1.1.1 (ref. 79; scaffold -q 60 -f 10000 -I 0.6 -remove-small) using the TAIR10 reference with hard-masked centromeres, rDNAs, telomeres and nuclear insertions of organelles to prevent misplacement of contigs due to reference bias75. All scaffolded assemblies were manually curated to specifically discard low-confidence centromere satellite-rich contigs or to invert contigs with satellite repeats at their edges, indicative of their correct orientation. These edits were implemented in the AGP files, which were converted to FASTA format with the RagTag agp2fa function79. To detect traces of residual heterozygosity, we aligned the original long reads to their corresponding chromosome scaffolds using pbmm2 v1.3.0 with the parameters align -sort -log-level DEBUG -preset SUBREAD -min-length 5000. Unmapped reads, as well as secondary and supplementary alignments, were filtered out using samtools v1.9 (view -b -F 2308 Chr1 Chr2 Chr3 Chr4 Chr5). The resulting BAM file was then analyzed with NucFreq v0.1 (-minobed 2) to assess genome-wide coverage of primary and secondary alleles80. AGP files, both before and after manual curation, as well as NucFreq plots, are available in the GitHub repository of this project.
Repeat annotation
Repetitive elements were annotated as described75. We ran RepeatMasker v4.0.9 (-cutoff 200 -nolow -gff -xsmall) using a custom library that included various consensus sequences for the CEN178 (ref. 81), 5S rDNA82, 45S rDNA83 and telomere repeats. We annotated tRNAs with tRNAscan-SE v2.0.6 (ref. 84) and TEs with Extensive de novo TE Annotator v1.9.7 (ref. 85; –step all –sensitive 1 –anno 1), a pipeline that combines several TE annotation tools (LTRharvest, LTR_FINDER, LTR_retriever, TIR-Learner, HelitronScanner and TEsorter)86,87,88,89,90,91,92,93. Finally, to understand the causes of contig breaks, we determined the type of repetitive element closest to each contig edge, considering the first 2 kb from each edge in contigs >10 kb.
Pannagram
Pannagram is a toolkit designed for reference-free pangenome alignment, annotation and analysis, as well as for generating diagrams46.
We represent the WGA as a matrix of corresponding positions, where rows represent accessions and columns represent homologous positions. The construction of the alignment is done in a reference-free manner (see below). However, to visualize the alignment in genome browsers, columns must be sorted in some manner, for example, to correspond to the TAIR10 sequence order. Then, columns of the pangenome are used as positions in the pangenome coordinate system.
To perform reference-free WGA, we developed a three-step pipeline. First, we use several accessions as references and build draft pairwise alignments between each and all other accessions. This process results in several reference-based matrices of corresponding positions. Next, we intersect these matrices, selecting only those columns that are present in all reference-biased matrices, which produces reliable and reference-independent correspondences. In the final step, we resolve unaligned sequences between blocks of corresponding positions using multiple sequence alignment tools. Once the reference-free alignment is complete, it can be sorted according to the desired order of accessions. In our case, we employ an alphabetical order, with the TAIR10 genome first.
For the pairwise alignments between a reference genome (not necessarily TAIR10) and another accession, the focal accession genome is divided into blocks of 5,000 bp, and each block is then mapped to the corresponding chromosomes of the reference genome using BLAST, with exactly one best hit retained for each block through this process. Next, the BLAST hits that are not in close proximity to each other in both genomes are removed. An additional BLAST search is performed to align corresponding unaligned sequences between remaining hits.
To resolve any unaligned blocks after the reference-randomization procedure, MAFFT94 is used. Blocks longer than 30 kb cannot be aligned within a reasonable time using MAFFT, so they are considered to be highly diverged. We found the final unaligned regions to be primarily associated with centromeric regions, rDNA clusters, telomeres and complex regions of multiple and long insertions and deletions, which are regions that are not of primary interest in this paper.
Given the WGA, SNPs can simply be output as sequence differences. However, sequence differences can arise from ambiguities in local alignment and do not necessarily correspond to SNPs (Supplementary Note 7). If we consider all sequence differences as SNPs, a pair of accessions differs at over 800,000 positions on average; however, if we restrict ourselves to isolated sequence differences, the number shrinks to 600,000.
Pangenome graphGraph construction
We constructed genome graphs for each of the five chromosomes using the PGGB pipeline29. First, we prepared the assemblies by splitting them into chromosomes and removing all unplaced contigs. To enforce linearity for simpler analysis and comparison, we used a modified version of accession 22001 with the genome rearranged to a consensus pan-genomic order (suffix: ‘f’). We added the TAIR10 reference genome to the graph to enable anchoring and presentation of results in a reference framework.
We executed the PGGB pipeline (downloaded on 25 January 2024) with the following parameters: -s 10000 -p 90 -n 27. PGGB consists of the following three methods: an all-against-all alignment with wfmash (v0.12.4-5-g0b191bb), graph induction using seqwish (v0.7.9-2-gf44b402) and two rounds of pangenome ordering (odgi v0.8.3-26-gbc7742ed) followed by normalization with smoothxg (v0.7.2-11-g9970e0d). The graph was used for analyzing the pangenome and synteny, as well as detecting variation using vg deconstruct95.
Similarity
We exploited graph properties to classify different levels of similarity between genomes. Nodes traversed in all accessions are labeled as core, nodes traversed in only one accession are private and all other nodes (>1 and <28 traversals) are shell (soft). Please note that nodes can be traversed multiple times by the same genome, which affects the total number of core nodes. Because each node contains a specific sequence, node count can easily be converted to the actual amount of sequence and respective genomic location.
Synteny windows
Every node in the graph can be translated to its exact position for each path. This direct connection allows us to create sliding window approaches for each sample/path using graph-based statistics. Here we used nonoverlapping windows of 300 kb and calculated the average similarity (see above) of these regions. This was performed for each graph and path independently and the results were represented in a heat map.
Saturation analysis
A saturation analysis was performed using a bootstrapping approach. In each iteration, we removed a specific number of paths from our graph and performed the same pangenome categorization as above (‘Similarity’). In addition, we added the total pangenome, which describes the total amount of sequence (core + shell + private sequence). We performed 20 different (unique) combinations for each size (number of genomes).
Deconstructing the graph
To achieve full insights into graph variation and cover all bubbles in the graph, VG deconstruct was run multiple times with each accession reference path once (vg deconstruct -a -e). After, the reported VCF (v1.54.0 ‘Parafada’) files were converted to a BED file with all important information provided. In addition, each chromosome was merged, and the genotype information was concluded and added. Bubbles were identified by the start and end positions, and all traversals within these bubbles were also reported. Scripts can be found in the repository.
sSVs and cSVs in the graphs were defined as follows:
All SVs represent indels, having one very small traversal (deletion) and a large one containing the SV sequence (insertion).
Bubbles were identified as sSVs if the bubble was shared by all accessions in the graph (here 28), and as cSVs if not. Traversals covering the insertion are at least 15 bp long and must exhibit high similarity (95% sequence). The deletion part of the bubble should be small, at most 5% of the length of the inserted sequence.
Most cSVs correspond to bubbles that have a complex structure and/or are sub-bubbles of larger bubbles.
General pangenome
To perform a reference-free pangenome analysis, we used genome graphs built separately for each chromosome. The complete graph contains 18.3 million nodes and 20.9 million edges, with a total size of 225 Mb, and has a mean compression rate of 6.75% across all chromosomes. Similar to other genome-wide analyses in this study, the large-scale reciprocal translocation in accession 22001 was masked to maximize linearity and increase resolution in the variation graph.
The mobile-ome
The mobile-ome refers to the collection of insertions and deletions that are likely to have occurred recently and are therefore not fixed in our sample. We hypothesize that each mobile event results in an SV, specifically a presence–absence polymorphism at the location of the insertion or deletion. Consequently, our initial approach involves extracting all presence–absence SVs and systematically decomposing them step by step. To distinguish between simple bi-allelic presence–absence polymorphisms (indels) and cSV, we analyzed the lengths of alleles within the SVs. We distinguish two types based on the similarity threshold s, with s = 0.9 in our case. We consider a simple indel as one that contains alleles of two length types—those that are shorter than (1 − s) of the SV length (absence allele) and those that are longer than s of the SV length (presence allele). The distinction between simple and complex presence–absence polymorphisms is partially a computational construct to filter SVs and simplify further analysis. Simple indels and complex presence–absence polymorphisms form a continuum, and by relaxing the similarity threshold (s < 0.9, in our case), some complex SVs become classified as simple indels. Additionally, there is a natural bias towards complex presence–absence polymorphisms. Consider a scenario with a simple presence–absence polymorphism where an indel occurs within the presence allele. If the presence allele was initially observed in only one accession, then the new event does not reclassify the initial region as not belonging to the simple presence–absence polymorphisms category. However, if the presence allele was observed in multiple accessions, the new event is likely to be reclassified as complex presence–absence polymorphisms. To simplify and clarify the analysis, we considered only the simple polymorphisms. To determine the known portion of the mobile-ome within indels, we conducted BLAST searches using pangenome consensus sequences of indels against known A. thaliana TEs, as well as against themselves. The indels that exhibited some similarity to known TEs were divided into the following groups: is complete—significant similarity to known TEs and can be classified as TEs themselves; contains complete—contained regions with similarity to known TEs, but also additional sequences; is fragment—contained only partially sequenced with similarity to known TEs; and contains fragment—partial coverage by BLAST hits of TE segments, but also additional sequences unrelated to known TEs.
We consider all these indels as parts of the mobile-ome. Indels without similarity to known TEs but showing nested similarities within the indel data set (where one sequence is a subsequence of another) were considered as potential candidates for new mobile-ome elements. To investigate their potential function, we obtained all six open reading frames within each of these indels. From each translated sequence, we selected either all continuous stretches without stop codons that were longer than 100 codons or the longest stretch that exceeded 30 codons without a stop. Subsequently, we performed a BLAST search using the obtained amino acid sequences against the NCBI protein database and classified the potential proteins into four categories. If the BLAST results for an sSV contained keywords related to TE, we assigned the sequence to the TE-like category. These keywords were ‘transcriptase’, ‘reverse’, ‘transpos’, ‘gag-’, ‘pol-’, ‘integrase’, ‘gag/pol’, ‘gagpol’, ‘retrovirus’, ‘RNA-directed DNA polymerase’ and ‘RNA-dependent DNA polymerase’. sSVs that only had BLAST hits with descriptions such as ‘hypothetical protein’, ‘unnamed protein product’, ‘uncharacterized protein’, ‘predicted protein’, ‘PREDICTED:’, ‘putative protein’ and ‘unknown’ were categorized as ‘undefined proteins’. Indels without any BLAST hits were classified as ‘no protein’. In all other cases, sSV was categorized as a ‘defined protein.’
Gene annotationPreliminary annotation
Gene annotation was mainly based on Augustus (v3.3.3)96. Augustus-predicted gene models were trained using parameters obtained from ‘hints’ from three different sources. First, we ran BUSCO (v4.0.1)97 with -m genome option. Second, the A. thaliana reference gene annotation was projected onto each genome using Liftoff98 with the -exclude_partial and -copies options. Third, the RNA-seq data for each accession were used—wiggle hints were generated using bam2wig and wig2hints, and EST hints were generated using bam2hints (all three tools provided by Augustus). Augustus was run with the following nondefault parameters:
–softmasking 1
–species=BUSCO_retraining
–gff3=on
–extrinsicCfgFile=Custom_Config
–hintsfile=Liftoff_hints
For every accession, the GFF3 output of Augustus was run through the Augustus-provided tool getAnno.pl to translate gene annotations into protein sequences. Finally, for each annotation, the Augustus output was combined and evaluated using augustus_GFF3_to_EVM_GFF3.pl (provided by EVidenceModeler99).
In addition to the Augustus-generated annotations, we used two types of independent evidence for gene models: from the SNAP de novo annotation tool100 and Cufflinks transcriptome assemblies101. Annotations produced by Augustus, SNAP and Cufflinks were combined and then subdivided into 1-Mb windows with 1-kb overlap using partition_EVM_input.pl (provided by EVidenceModeler). We ran EVidenceModeler with annotation GFF files, the assembly fasta file, the partitions and a weight matrix. We chose weights for each input based on their ability to recreate the Araport11 gene annotation. Running EVidenceModeler produced the final annotation compilation for each accession. We retained only the longest isoform for each gene using gffread102.
Reconciling annotations
To enable comparison between the independent annotations, we used the pangenome coordinate system, reconciling discrepancies using majority voting (Supplementary Note 6—‘Details about reconciling annotations and gene filtering’). Additionally, we compared the sequences of each gene across different accessions. If a gene showed significant variation because it was located in regions heavily influenced by SVs, we excluded it from the analysis. In total, 3,438 genes in our annotation were the result of splitting preliminary annotations and 1,020 were the result of merges. Lastly, we added 1,789 TAIR10 genes that had not been detected by our annotation pipeline (the likely reason for which is that our RNA-seq data only covered four tissues/stages) to our annotation. For these genes, the same pangenome coordinate approach was used to map the TAIR10 annotation of the 1,789 added genes into their annotations in other genomes. Our approach generated a total of 34,153 putative genes. For the details of annotation reconciliation and filtering, see Supplementary Note 6—‘Details about reconciling annotations and gene filtering’.
Ancestry analysis
All PC sequences from all accessions were compared using DIAMOND’s blastp module103 (version 2.0.11) against the A. lyrata MN47 proteome (version 2, GenBank: GCA_944990045.1), and the best hit was considered as the A. lyrata homolog. To avoid bias due to mis-annotated genes in the A. lyrata proteome, we further applied Liftoff v1.63 (ref. 98) to annotate all A. thaliana genes from all accessions on A. lyrata MN47 (v2, https://doi.org/10.6084/m9.figshare.22285444.v1) and A. lyrata NT1 (v2, https://doi.org/10.6084/m9.figshare.22293196.v1) assemblies. Next, each annotation group from A. thaliana was assigned to the A. lyrata homolog (by LiftOff or proteome similarity) that was common to at least 50% of its members, sharing at least 80% identity and covering at least 80% of the A. thaliana coding sequence. A. thaliana annotation groups were defined to be ancestrally relative to the A. lyrata gene if they were part of a colinear segment of at least two genes. To that end, all A. thaliana genes were ordered according to their relative position in the pangenome coordinate system. Each pair of consecutive genes in A. thaliana was assigned to the same colinear segment as its homologs in A. lyrata if the homologs were separated by fewer than six genes. The ancestral state was defined as ‘similar’ for cases where the genes from A. lyrata and A. thaliana were not part of the same colinear segment but shared at least 80% sequence identity over at least 80% of the length A. thaliana gene. Further details are available in Supplementary Note 6—under ‘Genes and TEs’ for TE analysis and ‘New genes’ for the origin of new genes.
Expression analysisRNA-seq read mapping and gene expression calculation
Raw RNA-seq reads from 7-day-old seedlings, 9-leaf rosettes, flowers and pollen54 were aligned either to the TAIR10 reference genome or the corresponding accession accession genome using STAR v2.7.1 (ref. 104) with the following custom options:
–alignIntronMax 6000
–alignMatesGapMax 6000
–outFilterIntronMotifs RemoveNoncanonical
–outFilterMismatchNoverReadLmax 0.1
–outFilterMismatchNmax 999
–outFilterMismatchNoverLmax 0.3
–outFilterMultimapNmax 1
–alignSJoverhangMin 8
–outSAMattributes NH HI AS nM NM MD jM jI XS
Read alignment statistics are provided in Supplementary Table 4. Expression levels were assessed using featurecounts from Subread v2.0.1 (ref. 105) on each RNA-seq sample with either the TAIR10 gene annotation or the accession-specific annotations from this study. The entire locus, including exons and introns, was used for expression estimates. Expression levels were normalized by calculating TPMs, which represent the number of read counts divided by the gene length in kilobases, and then dividing the total number of counts per kilobase for all genes by 1 million.
Mapping to TAIR10 versus the own genome
To determine whether the gene expression calculation was consistent between RNA-seq mapping in TAIR10 versus accession-specific genomes, we focused on the annotation groups with a one-to-one correspondence with an Araport11 gene. For each RNA-seq sample, we obtained the Pearson’s correlation coefficient between the number of exonic counts obtained from TAIR10 mapping and accession-specific mapping. We also determined the number of genes that were correctly or wrongly estimated using TAIR10 mapping. We called a gene ‘wrong’ if the counts in TAIR10 and the counts in its own genome differed by more than 30% (Ncounts_min/Ncounts_max ≤ 0.7). Only genes with at least six counts in either calculation were analyzed.
Chromatin immunoprecipitation followed by sequencing analysis
We used chromatin immunoprecipitation followed by sequencing (ChIP–seq) data from 6 accessions and sRNA-seq data from 14 accessions54. We used STAR104 to map ChIP–seq reads with these nondefault options:
–alignIntronMax 5
–outFilterMismatchNmax 10
–outFilterMultimapNmax 1
–alignEndsType EndToEnd
–twopassMode Basic
The ChIP–seq data were log2-normalized to input using bamCompare (deeptools package106) using
The ChIP–seq coverage was estimated using bedtools map-mean107. The ChIP–seq coverage was further normalized to obtain value range similarity across accessions. For this, we applied quantile-normalization using an R function:
function(x) { (x-quantile(x,.20)) / (quantile(x,.80) – quantile(x,.20)) }
which equalized the 20% and 80% quantile values of each ChIP–seq sample. After quantile-normalization, the replicated samples were averaged.
sRNA-seq analysis
We used sRNA-seq data for 14 accessions54. To process the sRNA-seq data, we trimmed the reads using cutadapt108: cutadapt -a AACTGTAGGCACCATCAAT –minimum-length 18. We then used STAR104 with the following nondefault options to map sRNA-seq reads to the corresponding genome:
–runRNGseed 12345
–alignEndsType Extend5pOfRead1
–alignIntronMax 5000 –alignSJDBoverhangMin 1
–outReadsUnmapped Fastx –outSAMmultNmax 100
–outSAMprimaryFlag AllBestScore
–outSAMattributes NH HI AS nM NM MD jM jI XS
–outFilterMultimapNmax 10
–outFilterMatchNmin 16
–outFilterMatchNminOverLread 0.66
–outFilterMismatchNmax 2
–outFilterMismatchNoverReadLmax 0.05
–outFilterIntronMotifs RemoveNoncanonicalUnannotated
–twopassMode None
We extracted 24-nt reads, calculated read coverage for each position of the genome using genomeCoverageBed (bedtools v.2.27.1), normalized it by the total number of uniquely mapped reads in each sample, and calculated 24-nt sRNA coverage for each locus of interest using bedtools map -mean function.
DNA methylation analysis
To estimate DNA methylation levels, we used published BS-seq data for 12 accessions53. After trimming with TrimGalore (https://github.com/FelixKrueger/TrimGalore) with –clip_r1 10 –clip_r2 15 –three_prime_clip_r1 10 –three_prime_clip_r2 10, reads for each accession were mapped to its corresponding genome with Bismark109 with –score_min L,0,-0.5 for a relaxed mismatch threshold and the –un –ambiguous parameters to obtain additional unmapped and multiply-mapping reads. Methylation was called as described110. CG, CHG and CHH methylation levels for genes and SVs in each accession were then calculated for each gene by focusing on all Cs in the specific context within the gene and calculating the ratio between the total number of methylated and unmethylated reads across all sites.
Mapping to TAIR10 versus own genome
To estimate reference bias, we mapped BS-seq data for all accessions to the TAIR10 genome and performed CG, CHG and CHH methylation level estimation in the same way as for own genomes. We then focused on annotation groups with a one-to-one correspondence with an Araport11 gene (the current annotation of the TAIR10 genome). We calculated Pearson’s correlation coefficient between the methylation level estimates obtained from TAIR10 mapping and accession-genome mapping. We also estimated the number of genes that were correctly or wrongly estimated using TAIR10 mapping. For each methylation context, we called a gene ‘wrongly estimated’ if the methylation level in TAIR10 and the own genome differed by more than 50% (methlevel_min/methlevel_max ≤ 0.5). For a more refined analysis of reference bias, see Supplementary Note 8.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.