
Enrollment rates
Between 28 September 2023 and 10 June 2024 (~8.5 months), a total of 2,125 newborns were enrolled. Recruitment letters were mailed to mothers of 42,434 newborns. We estimate approximately five enrollments for every 100 letters mailed (~5%). During the same period, 816 postpartum mothers were approached at UNC Hospital. Most (71%) were willing to learn about Early Check, and 183 (32%) enrolled their newborn during the encounter; we cannot estimate how many enrolled after their engagement with the recruiter. Few parents completed the decliners survey (n = 16). Reasons for not enrolling endorsed by three or more parents included concerns with DNA storage, worry about my child’s wellbeing, worry about my child’s privacy and not interested in research studies.
Participant characteristics
Compared to birthing mothers in North Carolina, self-report race/ethnicity data in Early Check indicated over-enrollment of white participants (58.8% versus 50.8%) (Table 1). Representation was lower among participants with both parents identifying exclusively as Black or Hispanic. However, significantly more Early Check participants reported two or more race/ethnicity categories than reported by the North Carolina State Center for Health Statistics (22.1% versus 2.9%).
Table 1 Characteristics of the first 1,979 newborns sequenced by Early CheckPanel selection
Panel 1 included 178 gene–condition pairs (169 unique genes) with high actionability during the first 2 years of life, whereas Panel 2 included an additional 29 gene–condition pairs. All enrolled newborns were screened for Panel 1 conditions. Additionally, 82.9% of parents opted for Panel 2. In comparison to mothers of white babies, mothers of babies reported as African American/Black (odds ratio (OR) = 0.37; P < .0001), Asian (OR = 0.61; P < .05), Hispanic/Latino or Spanish (OR = 0.43; P < 0.001) and Unknown/Not Reported (OR = 0.30; P < .01) were significantly less likely to choose Panel 2 screening.
Frequency of successful screening
Of 2,125 enrolled newborns, screening was completed for 1,979 (93%). Successful screening required a series of steps. First, the Early Check team identified and ‘matched’ the state NBS card with the consenting parent. Second, the team confirmed sufficient residual dried blood spot (DBS) material to punch. Third, the laboratory confirmed adequate DNA extracted for genome sequencing (GS). Participant attrition at each step is shown in Fig. 1. All consents that did not match an NBS card had discrepant information (for example, mother’s name did not match) and the consenting parent did not respond to requests for identity verification. One participant was administratively withdrawn after learning that the father provided consent on behalf of the mother, without the mother’s permission. Parents of newborns whose samples failed at any step were notified that screening was not completed. No significant differences in race/ethnicity were observed between newborns who were and were not sequenced successfully.
Fig. 1: Flow diagram of participant attrition from enrollment to results.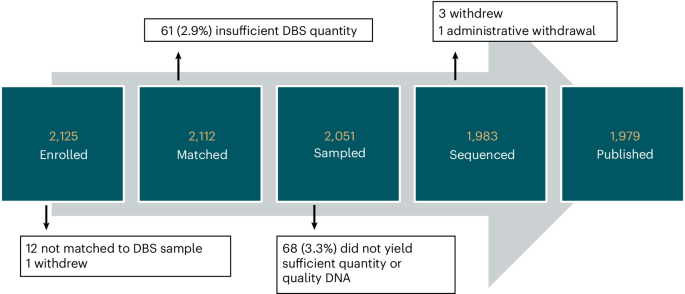
Enrolled: signed the consent. Matched: consent identifiers matched to an NBS card. Sampled: sufficient DBS sample remained on the NBS card to punch. Sequenced: successful sequencing from DBS punches. Published: participants reported in this paper.
Turnaround time
Median time from consent to return of results was 35 days (range 16–104 days) for negative results and 38 days (range 24–82 days) for positive results. The median time from consent to shipping samples to GeneDx was 11 days (range 1–63 days). The average time from sample arrival at GeneDx to completion of the results report was 13 days (range 6–40 days). The median time from completion of the results report, transfer of the report between institutions and availability of report in the Early Check portal was 5 days (range 1–58 days).
Screen-positive results
Fifty newborns (2.5%) received a positive screening result. One newborn had two positive results (SCN1A and MITF). Most positive results were from Panel 1 (n = 47), with four from Panel 2 (two SCN1A and two MECP2; Supplementary Table 1). The most common screen-positive results were G6PD deficiency and a pathogenic (P) variant (c.952G>A) in MITF associated with increased risk for melanoma15. Variants in G6PD accounted for nearly half of all screen positives, with a positive rate of 1.2%. The MITF gene was included in Panel 1 due to its association with Waardenburg syndrome, not melanoma risk16; therefore, the c.952G>A variant is considered an off-target result (phenotypic false positive). Excluding the 11 newborns whose only finding was the off-target c.952G>A variant in MITF, the overall positive screening rate was 2.0%. Excluding both MITF and G6PD screen positives, the overall positive screening rate was 0.8%.
Return of results
Several metrics were collected for each screen-positive result (Table 2). At the time of screening result disclosure, two newborns already had symptoms or a clinical diagnosis of the condition (COL2A1—Stickler syndrome, TMPRSS3—congenital hearing loss). Four parents were aware of a positive family history (F8—Hemophilia A, COL2A1—Stickler syndrome) or their carrier status (SLC26A4—Pendred syndrome, G6PD—G6PD deficiency) before enrollment. Screen-negative results (n = 1,929; 97.5%) were returned electronically.
Table 2 Results and follow-up metrics for 51 screen-positive results in 50 participantsConfirmatory molecular testing
Of 50 newborns with screen-positive results, 37 parents (74%) agreed to follow-up testing of their newborn and 32 (64%) provided newborn buccal samples for confirmatory molecular testing. One newborn had undergone clinical genetic testing for COL2A1—Stickler syndrome due to family history, so further confirmatory testing was unnecessary. Samples for follow-up testing were collected at home for 28 newborns and at UNC for four newborns. In all completed cases, the variant(s) identified on screening were confirmed. At publication, 17 participants (34%) had not completed molecular confirmation, mostly for nonurgent conditions (G6PD (n = 12); MITF (n = 3)).
Additional follow-up testing
Additional testing beyond molecular confirmation was recommended for seven newborns and completed for six (Table 2). Hearing evaluation confirmed the initial screen-positive result for two newborns (SLC26A4—Pendred syndrome, TMPRSS3—hearing loss). Other orthogonal testing modalities did not confirm any diagnoses. Concurrent biochemical testing for the newborn with IDUA variants demonstrated normal enzyme activity; urine glycosaminoglycan was not completed due to an unsatisfactory specimen. For three others, follow-up testing was normal or inconclusive; these newborns were deemed at risk to develop symptoms later in life (COL4A5—Alport syndrome, SCNN1B—pseudohypoaldosteronism, DUOX2—thyroid dyshormonogenesis).
Penetrance in infancy
Most infants with screen-positive results did not have signs or symptoms of the condition within the first few months of life. Three exhibited symptoms consistent with their molecular diagnoses: SLC26A4—Pendred syndrome, TMPRSS3—hearing loss and COL2A1—Stickler syndrome (Supplementary Table 1). Six infants had symptoms that were potentially associated with their molecular diagnoses. Five with G6PD deficiency had a history of neonatal jaundice, and one with a likely pathogenic (LP) variant in SCN1A had seizure-like episodes, although a normal EEG.
Comparison with standard state NBS
Three screen-positive results could have resulted in abnormal NBS due to their clear association with hearing loss (SLC26A4—Pendred syndrome, COL2A1—Stickler syndrome, TMPRSS3—hearing loss). However, only the newborn with variants in TMPRSS3 failed the NBS hearing screen. The newborns with SLC26A4 and COL2A1 variants were confirmed to have Pendred syndrome and Stickler syndrome, respectively, but did not have congenital hearing loss, consistent with natural history studies that indicate variable expression at birth17,18.
A newborn with a screen-positive result (DUOX2—thyroid dyshormonogenesis) had negative NBS despite having a homozygous P frameshift variant. Follow-up studies revealed normal thyroid function, and this result remains categorized as a ‘probable’ molecular diagnosis. Continued follow-up will be necessary due to the incomplete penetrance and variable expressivity described in this condition19.
Standard NBS results were reviewed for all 1,979 newborns screened by Early Check. Three newborns with confirmed diagnoses through standard state NBS were not detected by Early Check, indicating potential false negatives on gNBS. First was a newborn with sickle cell disease. Although initial gNBS was reported to the parents as negative, reinspection of the GS data revealed a homozygous P variant p.E7V in the HBB gene, and a revised, positive screening report was issued.
The second false negative was a newborn with late-onset Pompe disease. Standard NBS follow-up identified one P variant and one variant of uncertain significance (VUS) in the GAA gene. Since Early Check does not report VUS, this participant was assessed as a carrier and the result was not reported as screen positive. The third was a newborn with congenital hypothyroidism, which is most commonly not monogenic. Early Check did not screen for all possible monogenic causes. Thus, it is unclear whether this result represents a false negative result within the context of detectable monogenic forms of congenital hypothyroidism.
Molecular diagnosis and result classification
We applied study-developed rubrics (Table 3 and Fig. 2) to classify our screen-positive results (Table 4). Barriers to ‘definite’ molecular diagnoses included detection of LP (versus P) variants, inability to determine phase and potential variant misclassification. True positive or probable true positive was assigned to 28 results (55%). Four results (8%) were possible true positive/possible false positive and require longer follow-up.
Table 3 Early Check rubric for categorizing molecular diagnosesFig. 2: Rubric for categorizing screen-positive results.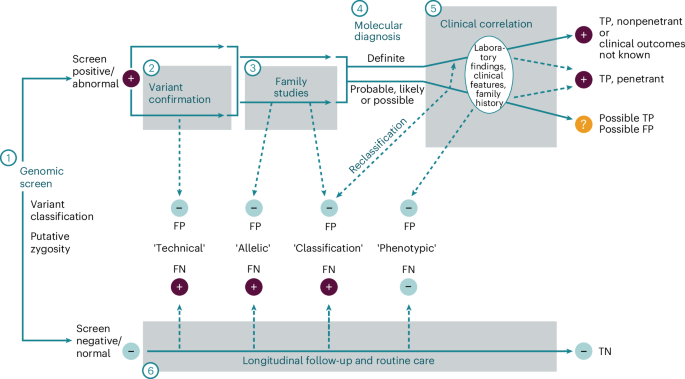
A genomic screening test must first have an established set of criteria that define a ‘positive’ or ‘negative’ screen (1), typically variant-level thresholds (for example, pathogenic/likely pathogenic versus VUS) and zygosity requirements for any given monogenic condition. Variant confirmation in a new sample using an orthogonal method (2) can identify rare cases of sample swap (estimated at 1:1,000 for clinical laboratory workflows) or artifactual variant calls (probability depending on the type of variant). Both types of laboratory errors could be considered ‘technical’ false positives that will happen at a somewhat predictable frequency. Family studies to determine de novo occurrence or to phase putative biallelic variants (3) may reveal that two variants reported as a positive screen are actually in cis and therefore not a disease-causing genotype, considered an ‘allelic’ false positive or, alternatively, may reveal inheritance of a heterozygous variant from an unaffected parent, potentially causing the clinical laboratory to downgrade the variant, considered a ‘classification’ false positive. A molecular diagnosis is thereby established (4) with varying degrees of confidence (Table 3). With general population screening, many people will initially be asymptomatic. Subsequent clinical correlation results in accumulation of laboratory findings, clinical features and family history (5). People with a definite molecular diagnosis are considered a true positive even if they never develop signs or symptoms of the condition (nonpenetrant). If clinical findings consistent with the putative molecular diagnosis emerge, then it can be confirmed as a true positive with penetrance established (though expressivity may vary). Sometimes, the phenotypic consequence of the variant(s) identified may end up being different than the intent of the screening program, such as hypomorphic variants with milder phenotypes, or variants having a different molecular mechanism that cause a phenotype not intended as part of the screen. These could be considered ‘phenotypic’ false positives, even though such findings may ultimately have clinical relevance. People with probable, likely or possible molecular diagnoses who remain asymptomatic over time make up a category in which the screening result could represent a false positive or a true positive (with incomplete penetrance and/or variable expressivity). Longitudinal follow-up and management of these people will be needed, thus creating additional burden with unclear benefit. Rarely, a person with a negative screen may develop symptoms of a monogenic disease over time, indicating a potential false negative (6). As with ‘technical’ false positives, a sample swap or low confidence variant call could result in a false negative. Similarly, a single heterozygous variant in a gene associated with a recessive condition could result in an ‘allelic’ false negative if the other variant was not detected, or a ‘classification’ false negative if the other variant was a VUS and therefore not reported. The category of VUS in general presents a challenge for screening in both recessive and dominant conditions, potentially resulting in ‘classification’ false positives and/or false negatives, depending on whether this category of variant is returned in a screening program. In some cases, disease manifestations may be due to locus heterogeneity (where the gene responsible for symptoms was not included in the screen) or because symptoms are caused by a nonmonogenic etiology; these would be ‘phenotypic’ false negatives. Each of these potential scenarios is unlikely in genomic screening due to the rarity of the screened conditions, but clinicians must remain alert to the possibility of a false negative as they are for any condition included in standard public health NBS. If a person remains asymptomatic over time, a negative becomes increasingly likely to be a true negative. FN, false negative; FP, false positive; TN, true negative; TP, true positive.
Table 4 Molecular diagnoses and result classification for the 50 screen-positive Early Check participants
Phenotypic false positives (n = 12, 24%) arose from detection of variants unrelated to the intended actionable condition, such as the MITF variant associated with melanoma risk. These diagnoses have clinical implications for participating newborns but were off-target findings. Parental testing revealed two maternally inherited variants in IDUA for one newborn, ruling out the initial molecular diagnosis of mucopolysaccharidosis type I. This result was classified as an allelic false positive. Four additional allelic false positives were due to detection of single heterozygous variants in genes associated with both autosomal dominant and autosomal recessive phenotypes and were more consistent with carrier status for the recessive condition.
Using our rubric, the sickle cell result that was initially reported as negative was classified as a technical false negative due to an oversight during a manual analysis step. This false negative may be correctable in the future by increasing automation support of sample analysis to increase throughput and reduce opportunities for error. The late-onset Pompe diagnosis detected on standard NBS but not gNBS is a classification false negative resulting from the stringency of the variant threshold used for reporting.
Clinical utility
The Early Check program demonstrated clinical utility by identifying actionable diagnoses for several families. One participant passed newborn hearing screening but was identified to have two P variants in SLC26A4. Auditory brainstem response performed at 15 weeks showed normal hearing sloping to mild/moderate sensorineural hearing loss. Repeat auditory brainstem response at 6 months showed bilateral mild sloping to severe mixed hearing loss. Brain MRI confirmed a diagnosis of Pendred syndrome. She received hearing aids at 8 months and is receiving speech therapy with good response to therapy goals. At 18 months she verbalizes 30–40 words and uses American sign language. Hearing loss continues to progress, with unilateral profound hearing loss. Assessment for cochlear implant is underway.
Two infants received actionable diagnoses of Stickler syndrome and TMPRSS3—hearing loss that were also identified clinically. Hearing loss in the infant with pathogenic variants in TMPRSS3 was initially attributed to congenital CMV until the molecular diagnosis was discovered. Parents of infants identified at risk for future morbidity received surveillance and management recommendations, along with connection to specialists, as needed.
Several screen-positive results from Panel 2 resulted in clinical actions, although were associated with greater uncertainty. One newborn with a de novo, previously reported, P variant in the MECP2 gene was diagnosed with Rett syndrome and referred to a multidisciplinary Rett Syndrome Clinic for management. Neurological and developmental assessments at 8 months were normal. The care team discussed the availability of trofinetide for children 2 years and older with the family; the family has expressed interest in interventional clinical trials for this or similar medications. Return appointment with the Rett Clinic team is planned for the second year of life to monitor development. One infant with an LP variant in SCN1A developed seizure-like episodes and was seen by neurology but subsequently lost to follow-up. Parents of the newborn with an LP variant in SCN1A that was downgraded by the sequencing laboratory to a VUS were given anticipatory guidance on seizures and are following with the pediatrician.