
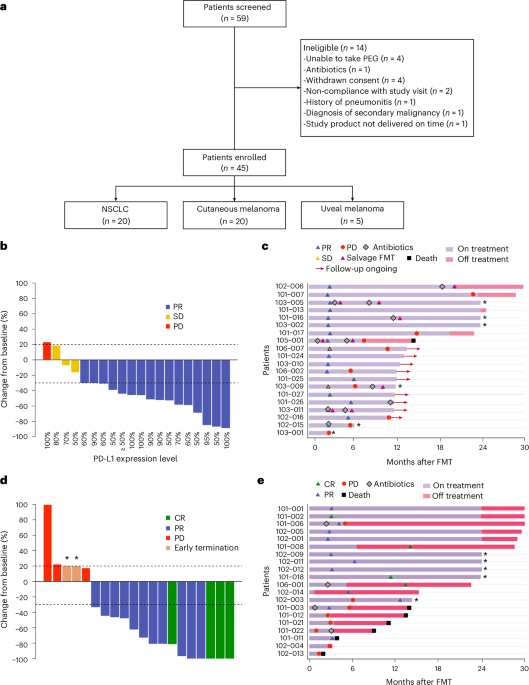
Study participants and clinical trial design
The FMT-LUMINate trial (NCT04951583) is a multicenter, open-label, phase 2 trial that included patients with advanced NSCLC, cutaneous melanoma and uveal melanoma who were being treated with first-line ICI therapy. Biological sex was considered in the study based on self-report. Gender was not considered in the study. Patients were 18 years of age or older with histologically confirmed diagnosis of NSCLC, cutaneous melanoma or uveal melanoma and measurable disease as per Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST v1.1). An Eastern Cooperative Oncology Group (ECOG) score of 0–2 was required. Previous treatment with palliative surgery or radiation was allowed. For the melanoma cohort, previous adjuvant anti-PD-1 was allowed provided the last dose occurred more than 6 months prior to enrollment. For the melanoma cohort, previous treatment with BRAF/MEK inhibitors was allowed. Major exclusion criteria included previous treatment with anti-PD-1 in the NSCLC cohort, a history of autoimmune diseases, active bowel disease, the use of daily corticosteroids >10 mg of prednisone or equivalent, symptomatic brain metastases or leptomeningeal disease. Patients were enrolled and treated at five academic centers in Canada: the Centre hopitalier de l’Université de Montréal (CHUM; Québec), the London Regional Cancer Program (LRCP; Ontario), the Centre hospitalier de l’Université de Québec (CHUQ; Québec), McGill University Health Centre (MUHC; Québec) and Lakeridge Health (Oshawa, Ontario). The research ethics board at each institution approved the conduct of the trial in accordance with current federal regulations, including the Canadian Food and Drug Regulations (C.05.001); the US Code of Federal Regulations (21 CFR Part 56); International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use Good Clinical Practice Guidelines; and the Declaration of Helsinki. The Unité d’Innovations Thérapeutiques at the CHUM was responsible for administrative functions, including the establishment of a DSMB, which provided safety oversight for the trial. Trial monitoring of the conduct of the trial and data management were performed by Ozmosis, Inc. All patients provided written informed consent and were able to withdraw consent at any time without compromising their cancer treatment. Patients were not financially compensated for their participation in this study.
Ethics approvals
The clinical trial and correlative analyses were approved by the CHUM Ethics Review Board and each participating center (ethics number MP-02-2022-10121/21.173). For culturomics analysis, this was conducted under project number 2025-12377 through biobank numbers MP-02-2018-7132/17.035 and 16.161.
Patient inclusion and exclusion criteria for enrollment
Inclusion criteria are summarized below:
1.
Age 18 years of age or older
2.
Confirmed histological diagnosis of advanced cutaneous melanoma, unresectable or advanced uveal melanoma or advanced NSCLC
3.
Stage IV or unresectable disease
4.
No prior anti-PD1 treatment with the exception of those who received adjuvant therapy (see point 5)
5.
For patients with newly diagnosed advance melanoma who relapsed after adjuvant immunotherapy, patients can be included in this study if they relapsed more than 6 months after their last dose of immunotherapy given in the adjuvant setting.
6.
For patients with NSCLC, tumor PD-L1 expression level ≥50%
7.
Evaluable disease as per immune-related RECIST (iRECIST) or RECIST
8.
ECOG performance status of 0−2
9.
Ability to ingest capsules
10.
Patients receiving systemic steroids at physiologic doses are permitted to enroll provided the dose does not exceed 10 mg prednisone daily or equivalent.
11.
Negative pregnancy test for women of childbearing potential
12.
Highly effective contraception (any method above 97% success rate) for both male and female patients throughout the study and for at least 60 days after last treatment administration if the risk of conception exists
Exclusion criteria are summarized below:
1.
Pregnant or breastfeeding or expecting to conceive or father children within the projected duration of the trial, starting with the prescreening or screening visit through 120 days after the last dose of trial treatment
2.
Current or recent (in the last 90 days) long-term exposure to high-dose oral or intravenous corticosteroids
a.
Patients who require intermittent use of bronchodilators, local steroid injections or short-term corticosteroids for any reason including, but not limited to, brain metastases treatment/prophylaxis are permitted to enroll at the discretion of the sponsor.
3.
Has a diagnosis of immunodeficiency (for example, HIV and transplantation) or receiving systemic steroid therapy (>10 mg prednisone daily or equivalent) or any other form of active immunosuppressive therapy
4.
Presence of a chronic debilitating intestinal disease (for example, malabsorption and colonic tumor)
5.
Use of probiotics during FMT. Probiotics must be discontinued a minimum of 24 hours before FMT administration, and patients are not permitted to take probiotics during the course of immunotherapy treatment.
6.
Use of antibiotics within 2 weeks of enrollment in the study
7.
Presence of absolute contraindications to FMT administration
a.
Toxic megacolon
b.
Severe dietary allergies (for example, shellfish, nuts and seafood)
c.
Active inflammatory bowel disease
8.
Expected to require any other form of systemic or localized anti-neoplastic therapy while on study. Palliative radiation therapy is permitted at the discretion of the sponsor.
9.
In the last year, has a known history of a malignancy requiring anti-neoplastic treatment:
a.
This time requirement does not apply to patients who underwent successful definitive resection of basal or squamous cell carcinoma of the skin, superficial bladder cancer and in situ cancers, including cervical cancer, breast cancer, melanoma or other in situ cancers.
10.
Symptomatic central nervous system metastases
11.
Leptomeningeal involvement (leptomeningeal enhancement on magnetic resonance imaging/computed tomography and/or positive cerebrospinal fluid cytology)
12.
Has an uncontrolled autoimmune disease that requires active immunosuppressive agents:
a.
Patients with vitiligo, type I diabetes, well-controlled hypothyroidism due to Hashimoto disease or resolved childhood asthma/atopy are not excluded.
13.
A history of (non-infectious) pneumonitis that required steroids or current pneumonitis
14.
Has serious concomitant illnesses, such as impaired cardiovascular function or clinically significant cardiovascular disease (uncontrolled congestive heart failure requiring treatment (New York Heart Association grade ≥3), uncontrolled hypertension, acute myocardial cardiac ischemia or unstable angina ≤2 months prior to study entry and severe cardiac arrhythmia) and active inflammatory bowel disorders
15.
Has an active infection requiring systemic therapy
16.
Patient has received a live vaccine within 4 weeks prior to the first dose of treatment:
a.
Seasonal influenza vaccines for injection are generally inactivated flu vaccines and are allowed; however, intranasal influenza vaccines (for example, Flu-Mist) are live attenuated vaccines and are not allowed.
17.
Has known psychiatric or substance abuse disorders that would interfere with cooperation with the requirements of the trial
FMT
Patients underwent a standard bowel preparation with 4,000 ml of PEG3350 liquid solution the evening before a single FMT using oral capsules. FMT capsules (now developed as LND-101) are produced using 80−100 g of feces per dose from screened healthy donors under the supervision of the Division of Infectious Diseases at St. Joseph’s Hospital in London, Ontario, Canada. Each dose of capsules contained the feces from a single donor. Major exclusion criteria for healthy donors included any known transmissible pathogen, history of medical illness, any history of major infection, such as COVID-19, monkeypox and hepatitis, and recent travel. A full list of inclusion and exclusion criteria was previously published23,51. Donor assignment was performed according to donor availability, without particular selection other than the screening requirements. Patients were required to consume 36–40 capsules under supervision and within 2 hours of defrosting the capsules, followed by a 30-minute period of observation. If a patient required antibiotics for an active infection throughout the study protocol, a ‘salvage’ FMT was performed within 14 days of the last dose of antibiotics52. Salvage FMT was not performed if the treating investigator deemed that the procedure would interfere with patient safety (that is, antibiotics administered during a severe adverse event (SAE) period). Patients were not required to undergo a second bowel preparation in the event of salvage FMT. In the event of Pneumocystis jirovecii prophylaxis, salvage FMT was not required.
Donor screening
Donor screening procedures are listed in Appendix 4 of the protocol and as previously published23.
Capsule preparation
FMT capsules were prepared according to an established protocol as described in our previous study23. In brief, feces donations (80–100 g) were processed separately without pooling by mixing in 0.9% normal saline and glycerol and were filtered using a stomacher bag. The filtrate was centrifuged, and the final sediment was mixed to incorporate residual liquid to allow pipetting into capsules as previously published23.
ICI therapy and assessments
ICI therapy was publicly funded. For the NSCLC cohort, patients received pembrolizumab at 2 mg kg−1 every 3 weeks, as per the standard of care. For the melanoma cohort, patients received ipilimumab 3 mgkg−1 intravenously plus nivolumab 1 mg kg−1 intravenously every 3 weeks for four cycles, followed by maintenance nivolumab 3 mg kg−1 every 2 weeks or nivolumab 6 mg kg−1 intravenously every 4 weeks. The first cycle of ICI was initiated within 7 days after FMT (Extended Data Fig. 1b). Therapy continued at the discretion of the treating oncologist until unacceptable toxicity, completion of 2 years, progression or death. Routine imaging was completed every 3 months for the first 2 years. Acceptable imaging included computed tomography or magnetic resonance imaging at the discretion of the treating oncologist who was encouraged to use the same modality of imaging throughout whenever possible. The assessment of treatment response was conducted as per RECIST v1.1 (ref. 53) and iRECIST54 when applicable at 3 months, 6 months and 12 months. Assessment of treatment response at all other timepoints was done by investigator-assessed RECIST. The ORR was defined as the proportion of patients with a complete response or a partial response and designated as ‘R’ for the translational studies. All other patients (the proportion with stable disease or progressive disease as best response), were designated as ‘NR’.
Data and sample collection
At each clinic visit, patient data, to include demographics, clinical and imaging assessments and adverse event recording, were collected per protocol using a secure, password-protected, electronic data capture web-based tool (Medidata Rave Unified Life Science Platform). Patient fecal and blood samples were collected at the following timepoints: before FMT (baseline), the first cycle of ICI therapy (1 week) and the second cycle of ICI therapy (1 month), 2 months, 3 months, 6 months, 9 months and 1 year. A complete clinical assessment was conducted at each visit with routine lab work per standard of care. Patients were permitted to withdraw consent at any time with no impact on treatment, although analysis of previously obtained samples was retained.
Assessment and management of AEs
FMT and ICI-related toxicities were evaluated using the Common Terminology Criteria for Adverse Events version 5.0 (CTCAE v5.0) grading scale55. Toxicities were attributed to FMT only if the investigator deemed the AE to be related to FMT and if the AE occurred before the first cycle of ICI. AEs were managed by the treating investigator in accordance with routine clinical practice guidelines. Any patient who experienced an SAE had ICI therapy suspended until resolved or discontinued at the discretion of the treating investigator. All AEs were reviewed by the trial sponsor to ensure consistency in attribution and grading.
Outcomes and sample size
The primary outcome of the study was the ORR in the NSCLC cohort, defined as the proportion of patients whose best objective response was either a complete response or a partial response. Best objective response was determined as the best response recorded from the first dose of study treatment until the last tumor assessment prior to initiation of subsequent therapy.
Secondary outcomes included DCR, defined as the proportion of patients achieving complete response, partial response or stable disease lasting more than 6 months; ORR in the melanoma cohort; and safety of FMT in combination with ICI. Additional secondary outcomes to be reported subsequently include PFS and OS at 12 months in both the NSCLC and cutaneous melanoma cohorts. Microbiome engraftment was evaluated by acquired donor–host similarity using the Bray–Curtis dissimilarity index.
Each cohort was analyzed independently, as outlined in the study protocol, given their differing disease biology and treatment context.
Sample size was determined as follows: for the NSCLC cohort, assuming that the ORR rate is 39% (null hypothesis)3, a sample size of 18 patients has 80% power to detect an ORR of 64% (alternative hypothesis) using a one-sided binomial test with 0.10 level of significance. Because the prespecified primary endpoint of the study was ORR in the NSCLC cohort, there was no prespecified sample size calculation for the cutaneous melanoma cohort.
The present paper focuses on the NSCLC and cutaneous melanoma cohorts given prespecified primary and secondary endpoints in these groups outlined in the study protocol. Given that the uveal melanoma cohort was explicitly defined to be an exploratory cohort, the uveal melanoma cohort results will be reported separately.
Due to the COVID-19 pandemic and widespread disruptions to clinical trial operations56,57,58—including delayed site activations and reductions in outpatient oncology services—accrual to the trial was slower than anticipated. The protocol was amended to a total target of 20 patients, enabling evaluation of the primary and key secondary endpoints while maintaining feasibility under pandemic-related constraints.
Metagenomics analysisSequencing and processing
A total of 199 fecal samples collected from 39 patients enrolled underwent whole-genome shotgun sequencing. Stool aliquots were preserved at –80 °C in DNA/RNA Shield Buffer (Zymo Research) until processing. DNA extraction was performed using the DNeasy PowerSoil Pro Kit (Qiagen), following the manufacturer’s protocol. Sequencing libraries were prepared using the Illumina DNA Prep (M) Tagmentation Kit (Illumina), following the manufacturer’s instructions, and purified with a 0.7× ratio of Agencourt AMPure XP beads. Sequencing was conducted on a NovaSeq 6000 S4 flow cell (Illumina) at the University of Trento’s in-house sequencing facility (Trento, Italy). Raw reads were processed through a quality control pipeline available at https://github.com/SegataLab/preprocessing. In brief, reads were filtered out if they had low quality (Q < 20), were shorter than 75 bp or contained two or more ambiguous bases. Host-derived reads (human genome hg19 and Illumina spike-in phiX174) were also removed. On average, each sample yielded approximately 48 million high-quality reads. Five samples failed internal quality control and were excluded from downstream analyses. Taxonomic profiling was carried out using MetaPhlAn 4 (database version vJun23_CHOCOPhlAnSGB_202307). Of the 194 samples that passed quality control, 12 were excluded from the downstream analysis: 10 of excluded samples occurred during unscheduled visits, and two excluded samples were collected after salvage FMT. Because the salvage FMT typically involved a different donor, all samples occurring after the patient received salvage FMT were excluded.
Statistical analysis for shotgun metagenomics sequencing
Microbiome sequencing data were processed and analyzed using the phyloseq package (version 1.50.0). Taxonomic assignments and abundance tables were imported into a phyloseq object, including corresponding sample metadata. α-Diversity analysis was performed using the vegan package (version 2.7.1), calculating richness and Shannon index. Principal component analysis was done with the packages prcomp (version 4.4.2) and factoextra (version 1.0.7), visualizing the variance explained by the principal components and the top contributing taxa. For the donor clusters unsupervised analysis, one sample from cluster A was removed from the visualization but included in the cluster calculation. PERMANOVA analysis was performed with 999 permutations using the ‘adonis’ function from the vegan package. Strain sharing between the patients and donors was calculated using StrainPhlAn4 with an in-house database for strain identification as previously published. Bray−Curtis dissimilarity was calculated between the samples and their corresponding donor samples using the ‘distance’ function from phyloseq. These distances were then adjusted to their corresponding baseline sample to observe the fold change over time. Linear discriminant analysis (LDA) effect size (LEfSe) was performed to identify microbial taxa that differentiate between subject groups according to their response, cohort or toxicity development using the package yingtools2 (version 0.0.1.184). We filtered taxa to be in at least 10% of the samples (prevalence cutoff of 5%). Features with LDA scores greater than 2 and P < 0.05 were retained. Taxonomic abundance patterns between groups were visualized via heatmaps generated by the ComplexHeatmap package (version 2.22.0). The analysis was based on differentially abundant taxa between conditions using a Wilcoxon rank-sum test.
Strain-level analysis
The strain-level analysis to assess strain engraftment was performed following the procedure previously published23. Phylogenetic trees were generated for each SGB with StrainPhlAn4. The thresholds on normalized phylogenetic distances to define same-strain pairs were determined for each SGB separately by comparing the distances between pairs of longitudinal samples to the pairs of unrelated samples in the corresponding phylogenetic tree, and their separation was maximized with Youden’s index. Strain engraftment of an SGB was detected if the donor sample had the same strain of the SGB as the post-FMT sample.
Calculation of SGB categories
For all patients with available corresponding donor shotgun metagenomic sequencing data, SGBs in each sample were categorized as follows: ‘Baseline’ for the SGBs unique only to the patient at their baseline sample; ‘Donor’ for the SGBs present in the sample that matched unique SGBs from the respective donor sample but were not found in the baseline sample; ‘Both’ for unique SGBs that were found in both the baseline sample and the matching donor sample; ‘New’ for SGBs not present in either the ‘Donor’ or the ‘Baseline’; and ‘Lost’ when SGBs that were identified at baseline were no longer present in the sample. To calculate the proportion of lost SGBs, we divided the ‘Lost’ SGBs by the total amount of SGBs present at baseline.
Pathway analysis from shotgun metagenomics sequencing
Shotgun metagenomic samples were also processed with the HMP Unified Metabolic Analysis Network (HUMAnN3) pipeline (version 3.8). We evaluated the stratified pathway abundances from the MetaCyc database. Pathways with zero total abundance across all samples were filtered out. Differential abundance analysis between baseline and post-FMT was performed using DESeq2 (version 1.46.0).
BiomScope pipelinePipeline description
Gene abundance profiling was performed using both the 10.1 million-gene integrated catalog of the human gut microbiome59 and the 8.4 million-gene integrated reference catalog of the human oral microbiome60. Filtered high-quality reads were rarefied to 20 million by seqtk and then mapped with an identity threshold of 95% of each catalog using Bowtie (version 2.4.4). A gene abundance table was generated with BiomScope software61 through a two-step procedure. First, the uniquely mapped reads (reads mapping to a single gene in the catalog) were attributed to their corresponding genes. Second, shared reads (reads that mapped with the same alignment score to multiple genes) were attributed according to the ratio of their unique mapping counts. For quantification of species, gene abundance was normalized using the fragments per kilobase of transcript per million mapped reads strategy (normalization by the gene size and the number of total mapped reads reported in frequency), and species abundance was determined from the average abundance of the first 100 core genes of each species provided in the annotation of the each million-gene catalog. Species with fewer than 10 core genes detected were counted with an abundance equal to zero. Species are reported as Metagenomic Species Pan-genomes (MSPs) that were defined and described previously62.
Species retention
Species were first categorized for each patient according to their presence status in baseline timepoint of patient and donor, defining the following categories: both, only patient, only donor and new categories. Species never seen at any timepoint were discarded. Species were then grouped by category and filtered for at least 10 observations—for example, seen in one of the categories for at least 10 patients. The proportion of patients with the species present at a given timepoint is called the species retention. The curve displaying species retention was obtained as the median of all retention values of the different species in a specific category, with 95% confidence intervals for median. P values were calculated using the Mann−Whitney−Wilcoxon test.
High-throughput culturomics
High-throughput culturomics was done as previously performed63. A total of 0.3 g of stool from enrolled patients was resuspended in 1 ml of sterile 1× PBS. From this suspension, seven serial dilutions were prepared, and 50 µl of each dilution was plated onto 5% Columbia agar supplemented with sheep blood (COS; Thermo Fisher Scientific). Plates were incubated aerobically and anaerobically in an anaerobic chamber (5% H2, 5% CO2 and 90% N2) at 37 °C for up to 72 hours. Anaerobic conditions were achieved using sealed Zip bags (Becton Dickinson) containing GasPak anaerobic generators (Becton Dickinson). In parallel, enrichment cultures were performed by inoculating 200 µl of stool suspension into BD BACTEC Lytic/10 Aerobic (Becton Dickinson) for aerobic conditions. For anaerobic conditions, BD BACTEC Lytic/10 Anaerobic/F (Becton Dickinson) and Yeast Casitone Fatty Acids Broth with Carbohydrates (YCFAC) were used. All broth cultures were supplemented with 5% defibrinated sheep blood and 5% filtered rumen fluid (0.22 µm). At multiple timepoints (24 hours, 3 days, 7 days, 10 days, 15 days, 21 days and 30 days), serial dilutions from enrichment broths were plated on COS agar and incubated under the same aerobic and anaerobic conditions. After incubation, isolated colonies were subcultured onto fresh COS agar and incubated at 37 °C for an additional 72 hours. Colonies were then purified and subjected to identification. Bacterial identification was performed using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS). For each isolate, a double spot was deposited onto a 96-spot MSP target plate, overlaid with 1 µl of a saturated α-cyano-4-hydroxycinnamic acid matrix solution (prepared in 50% acetonitrile and 2.5% trifluoroacetic acid) and allowed to dry. Spectra were acquired using a MicroFlex LT/SH mass spectrometer (Bruker) and analyzed via FlexControl version 3.4 and MALDI Biotyper Compass version 4 software. Identification was achieved by comparison to the Bruker MBT Compass BDAL reference library and an in-house spectral database. Colonies yielding identification scores higher than 1.9 were considered reliably identified at the species level. Isolates with scores lower than 1.9 were further analyzed by whole-genome sequencing for taxonomic assignment. Bacterial species detected in patient samples at baseline were compared to those identified 1 month after FMT. Due to lack of donor samples available for culturomics, we used the corresponding donor shotgun metagenomics sequencing sample. Species detected at baseline but absent at the 1-month post-FMT timepoint were classified as lost. Conversely, bacterial species present in both the donor sample and the recipient 1 month after FMT—but not detected in the patient at baseline—were defined as engrafted from the donor. Engraftment from the donor versus lost from baseline was compared using the non-parametric Mann–Whitney U-test using GraphPad Prism (version 10.4.2) software.
Metabolomic analysis
Fifty microliters of plasma was mixed with 500 μl of ice-cold extraction solution (methanol:water, 9:1, v/v, at −20 °C) containing isotopically labeled internal standards. The mixture was vortexed for 5 minutes at 2,500 rpm to ensure thorough homogenization and efficient extraction of endogenous metabolites. Samples were then centrifuged at 15,000g for 10 minutes at 4 °C. The resulting supernatants were collected and aliquoted into multiple fractions to be analyzed by different liquid chromatography coupled with mass spectrometry (LC−MS)64. Targeted detection of polyamines and bile acids was performed by LC−MS/MS using a 1290 Ultra-High-Performance Liquid Chromatography (UHPLC) system (Agilent Technologies) coupled to a 6470 triple quadrupole mass spectrometer (QQQ 6470) (Agilent Technologies). For polyamine quantification, 10 μl of extract was injected into a Kinetex C18 analytical column (150 mm × 2.1 mm, 2.6-μm particle size; Phenomenex), equipped with a C18 guard column (5 mm × 2.1 mm). The column was maintained at 40 °C using a Peltier oven. Chromatographic separation was achieved using a binary mobile phase consisting of (A) water with 0.1% heptafluorobutyric acid (HFBA) and (B) acetonitrile with 0.1% HFBA, both freshly prepared. The initial mobile phase composition was 95% A and 5% B, followed by a linear gradient to 30% B over 7 minutes. The column was washed with 90% B for 2.25 minutes and then re-equilibrated with 5% B for 4 minutes. The autosampler was maintained at 4 °C. Mass spectrometry parameters included gas temperature of 350 °C, gas flow rate of 12 l min−1 and capillary voltage of 2.5 kV. For bile acid analysis, 10 μl of sample was injected onto a Column Poroshell 120 EC-C8 1,200 bars (100 mm × 2.1 mm, 1.9-μm particle size; Agilent Technologies), protected by an XDB-C18 guard column (5 mm × 2.1 mm, 1.8 μm). The column was also maintained at 40 °C. The mobile phase consisted of (A) water with 0.2% formic acid and (B) acetonitrile:isopropanol (1:1, v/v), both freshly prepared. Initial conditions were 70% A and 30% B, followed by a shift to 38% B over 2 minutes, maintained for an additional 2 minutes. A rapid gradient from 38% to 60% B was applied over 30 seconds. The column was washed with 98% B for 2 minutes and re-equilibrated at 30% B for 1.5 minutes. Autosampler temperature was kept at 4 °C. Instrument parameters were set with gas temperature of 310 °C, gas flow rate of 9 l min−1 and capillary voltage of 4.5 kV. In addition, pseudo-targeted metabolomic profiling was performed using ultra-high-performance liquid chromatography coupled with high-resolution accurate mass spectrometry (UHPLC−HRAM) on a Dionex U3000 UHPLC system coupled to a Q-Exactive Orbitrap mass spectrometer (Thermo Fisher Scientific), as previously described65,66. All targeted treated data were merged and cleaned with a dedicated R package (@Github/Kroemerlab/GRMeta). We applied the ComBat function from the sva R package (sva version 3.52.0) to correct for inter-batch effect. Batches were divided into two groups. The batch correction model included a design matrix preserving the biological signal from key clinical covariates. Clinical features associated with samples included age, gender, body mass index (BMI), tumor type and time of first dose of ICI. ComBat was applied on log10-transformed AreaQCCorr values.
Statistical analysis for metabolites
Metabolite longitudinal data were analyzed using generalized additive models (GAMs) with the mgcv R package (version 1.9-3) using the ‘gam’ function. Each metabolite was fitted with a GAM model that included the two groups (R and NR) present in the study. Before applying any model, data were cleaned by removing individuals with only one timepoint of analysis and duplicated samples. After cleaning the dataset, this metabolite analysis comprised 100 samples from 35 patients, each having at least two timepoints sampled (from two to four). Data normalization was performed before model fitting. Metabolite intensities were transformed using square root normalization with the ‘sqrt’ function from R base (version 4.5). Individual GAMs were fitted for each metabolite with the following model structure:
a.
Smooth terms for time with group-specific evolution over time using factor smooth interactions
b.
Random effects for individual patients
c.
Tweedie distribution to account for the data distribution characteristics
d.
Restricted maximum likelihood (REML) estimation method
Time was modeled with penalized splines. The effective degree of freedom (edf) indicates the complexity of the time curve. In the fitted models, edf values excedeed 1, which is consistent with a nonlinear pattern. Model performance was assessed by calculating root mean squared error to evaluate model accuracy deviance explained and R2 to assess how much the model explained the signal variance, along with residual analysis and concurvity assessment. Temporal evolution was evaluated from the fitted group-specific time smooth across the entire observation window. Reported P values are the significance tests for these time smooths, assessing the null hypothesis of no within-group time effect (flat curve). Temporal evolution was evaluated through model outputs and P value. Between-group differences were calculated using the ‘difference_smooths’ function from the gratia package (version 0.10.0) to compute differences between R and NR smooth curves with 95% confidence intervals. Figures displaying temporal evolution were created using smooth estimates calculated with the ‘smooth_estimates’ function from the gratia package. Both trajectory and difference plots were generated using the ‘ggplot’ function from the ggplot2 package (version 3.5.2) and combined using the ‘plot_grid’ function from the cowplot package (version 1.1.3). P values were displayed using the ‘ggsignif’ function from the ggsignif package (version 0.6.4). Data manipulation was performed using the dplyr package (version 1.1.4) and tidyverse (version 2.0.0). All analyses were conducted in R (version 4.5).
Bio-Me qPCR assay
We used Precision Microbiome Profiling (PMPTM) (Bio-Me), which is a validated qPCR method for analyzing the gut microbiome composition, based on TaqMan technology on the OpenArray format (Thermo Fisher Scientific). This assay targets 108 bacterial species and subspecies (107 bacteria and one fungal species). Standard curves for the assays were created using reference materials quantified by fluorescence (Thermo Fisher Scientific, Quant-iT PicoGreen dsDNA Reagent). The reference materials were acquired from the Leibniz Institute DSMZ or the American Type Culture Collection. Standard curves for each qPCR assay were used to convert the quantification cycle (Cq) value into number of genomic copies per microliter of sample; this number was transformed into normalized absolute quantification. qPCR was performed on 220 samples in total (n = 181 patients and n = 39 donors), and only patient samples with corresponding SGB category information were included in the final analysis (n = 104 samples). Quantitative PCR and metagenomics relative abundance were visualized using the ComplexHeatmap package in R, and a Kendall score Kendall’s τ was computed using the ‘cor.test’ function in the ‘stat’ package using pairwise complete observations. Spearman′s r correlations were performed using GraphPad Prism.
Olink proteomics
Multiplex high-throughput proteomics was performed using the Immuno-Oncology Panel (Olink) and analyzed per the manufacturer’s instructions. All Olink data are reported as linearized normalized protein expression (NPX), per the manufacturer’s instructions. Olink data were visualized in R using the OlinkAnalyze package using ‘olink_umap_plot’ using manyfold approximations and projections using uniform manifold approximation and projection (UMAP) and ‘olink_volcano_plot’. For volcano plots, Benjamini−Hochberg-corrected P values less than 0.05 were considered statistically significant.
Quantification of 16S PCR
Quantitative real-time PCR was performed to assess the relative abundance of total bacterial DNA in stool samples by targeting the V6 region of the 16S rRNA gene. The primer pair used was 891F (5′-TGGAGCATGTGGTTTAATTCGA-3′) and 1033R (5′-TGCGGGACTTAACCCAACA-3′)67. For each reaction, 400 ng of extracted DNA was combined with 500 nM of each primer and 1× qPCRBIO SyGreen Blue Mix Hi-ROX (PCR Biosystems). Amplification was carried out using a real-time PCR system, and threshold cycle (Ct) values were obtained. Bacterial load was estimated by comparing sample Ct values to a standard curve generated using serial dilutions of Escherichia coli genomic DNA, allowing for the approximation of bacterial DNA concentration in ng μl−1. Non-parametric Mann−Whitney−Wilcoxon test to compare SGB loss groups and two-way ANOVA were performed in GraphPad Prism.
Peripheral blood mononuclear cell immuno-phenotypingSpectral flow cytometry staining and acquisition
Aliquots of cryopreserved peripheral blood mononuclear cells (PBMCs), each containing 5 × 106 cells preserved in fetal calf serum (FCS) with DMSO, were quickly thawed, gently washed and resuspended in FACS buffer consisting of 1× PBS supplemented with 5% FCS and 2 mM EDTA. To exclude non-viable cells, samples were incubated with the LIVE/DEAD Fixable Blue Dead Cell Stain Kit (Invitrogen) according to the manufacturer’s protocol. Surface staining was performed using fluorochrome-conjugated antibodies sourced from BD Biosciences, R&D Systems, MBL, BioLegend, Miltenyi Biotec and Cytek (see reagent list for detailed panel composition; Supplementary Table 3). Data were acquired with a Cytek Aurora 5-laser spectral analyzer, and data analysis was performed using FlowJo version 10.8.1 software.
Manual gating and high-dimensional flow cytometry analysis
Flow cytometry analysis of PBMCs followed a sequential gating strategy that excluded debris, cell doublets and non-viable cells, ultimately selecting for viable CD45+ immune cells. The resulting CD45+ live-cell populations were exported as new .fcs files using FlowJo version 10.8.1, and these curated datasets served as input for UMAP and Single-cell Cytometry Annotation Network (Scyan) analyses.
Algorithms for dimensionality reduction: UMAP analysis
The UMAPs were computed and displayed using the Scanpy package68 on alive CD45+ cells.
Scyan automatic annotation
Cell type annotations were automatically performed using Scyan69 a biology-driven model that leverages prior knowledge of cell types. This approach allowed us to identify nine main populations: basophils, natural killer cells, B cells, monocytes, dendritic cells, CD4+ T cells, CD8+ T cells, double-positive T cells and double-negative T cells. Subsequently, for each patient, separate .fcs files were generated for each of these nine populations. The percentages of all subpopulations were then determined through manual gating using FlowJo version 10.8.1.
Murine experiments
All animal studies were approved by the Institutional Animal Care Committee at the Centre de Recherche du Centre Hospitalier de l’Université de Montréal (CRCHUM) and carried out in compliance with Canadian Council on Animal Care guidelines (ethical protocol C22017Br). Murine experiments were conducted using 7-week-old female wild-type C57BL/6 SPF-reared mice obtained from Charles River Laboratories. Mice received 3 days of antibiotics solution containing ampicillin (1 mg ml−1), streptomycin (5 mg ml−1) and colistin (1 mg ml−1) (Sigma-Aldrich), which was added to the sterile drinking water of mice before FMT. Three consecutive FMTs, using patient feces (ethics approval was obtained (MP-02-2022-10121/21.173, MP-02-2018-7132/17.035 and 16.161)), were performed at day −15, day −14 and day −13 (Fig. 4i). FMT was performed by thawing fecal material, and 200 µl of the suspension (100 mg ml−1) was then transferred by oral gavage. An additional 100 µl was applied on the fur of each animal.
Two weeks after the first FMT, mice were implanted subcutaneously with 0.8 × 106 MCA-205 cells. When tumors reached 25−35 mm2 in size or 35−45 mm2 in size for anti-PD-1 monotherapy and anti-PD-1 plus anti-CTLA-4 experiments, respectively, mice were treated four times intraperitoneally every 3 days with anti-PD-1 monoclonal antibody (250 μg per mouse; Bio X Cell, clone RMP1-14) or anti-PD-1 monoclonal antibody plus anti-CTLA-4 monoclonal antibody (100 µg per mouse; Bio X Cell, clone 9D9). Additionally, the mice received a 200-µl gavage of NaCl or bacterial cocktail group starting 3 days after the tumor implantation and at each treatment. Tumor sizes were measured with manual calipers. The mice were euthanized 2 days after the last treatment. Experimental groups were compared using the Mann−Whitney−Wilcoxon test in GraphPad Prism.
Bacterial cocktail group preparation
For murine experiments, bacterial cocktails were prepared as follows. Bacterial strains used in the murine experiments were isolated from patients at baseline enrolled in the FMT-LUMINate clinical trial and stored at –80 °C. Bacteria were cultured from frozen stocks on fastidious anaerobe agar plates (Thermo Fisher Scientific, PB0225A) for 48 hours at 37 °C under anaerobic conditions. Prior to cocktail formulation, the identity of each isolate was confirmed by MALDI-TOF MS. After identification, bacterial colonies were collected, resuspended in sterile NaCl and adjusted to an optical density of 1.0, corresponding to approximately 1 × 109 colony-forming units (CFU) per milliliter.
Cell culture, reagents and tumor cell lines
MCA-205 fibrosarcoma cells, class I MHC H-2b syngeneic cell lines for C57BL/6 mice, were used for this study and obtained from Jonathan Stagg’s laboratory. The cells were cultured at 37 °C in the presence of 5% CO2 in RPMI 1640 containing 10% FCS, 2 mM L-glutamine and 100 UI ml−1 penicillin−streptomycin. All reagents were purchased from Gibco-Invitrogen. Cell lines were checked for mycoplasma using PlasmoTest Mycoplasma Detection Kit (InvivoGen).
Statistical analysis
All statistical analysis and visualization were done in R or GraphPad Prism (version 10.4.2). P values were considered statistically significant if P < 0.05, and all P values were two-sided.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.