
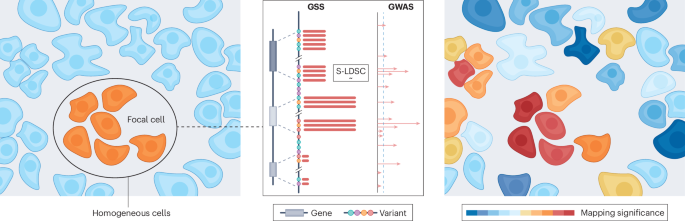
Genome-wide association studies (GWAS) have uncovered numerous genetic variants associated with complex traits. However, a critical gap remains in pinpointing the specific cells in which variants exert their effects, particularly within the tissue context. Previous methods have combined single-cell RNA-sequencing data with GWAS summary statistics to identify trait-associated cells or cell types, but these approaches fail to reveal how these cells are spatially organized within tissues. We developed gsMap, an efficient computational framework that integrates spatial transcriptomics (ST) data with GWAS summary statistics to pinpoint trait-relevant cells in situ.
As human ST data are limited, gsMap has been primarily applied to ST data from model organisms. Its application to mouse embryonic ST data showed that gsMap can recapitulate known region–trait associations, supporting its potential for broader use and cross-species mapping. For example, intelligence quotient was linked to brain regions, mean corpuscular haemoglobin concentration to liver regions, and height to multiple tissues, with the strongest signals in cartilage and its primordium.