
Introduction of centenarian COL25A1 SNP in C. elegans ortholog col-99
In 2001, Puca and colleagues performed a linkage study in centenarian sibling pairs to identify genetic loci associated with exceptional longevity5. Interestingly, they identified a 3 cM locus on chromosome 4 that was strongly associated with longevity5. However, the human genome was not yet sequenced at the time of publication, and this region had not been characterized. Therefore, we investigated this locus and found 108 ensemble features, including 33 protein-coding genes, 14 lincRNA, 2 miRNA, 2 snoRNA, and 9 snRNA (Figs. 1a, S1a, Supplementary Table 1). To identify causally implicated protein-coding genes, we cross-referenced genes to identify those with longevity-associated SNPs in centenarian GWAS or with studies showing that altering gene function would increase lifespan in any organism (Fig. S1a, Supplementary Table 1). Strikingly, we found a collagen gene, COL25A1, which was previously shown to be significantly associated with human healthy aging (“Wellderly phenotype”), but without mechanistic validation9.
Fig. 1: Mapping longevity SNP from humans in C. elegans.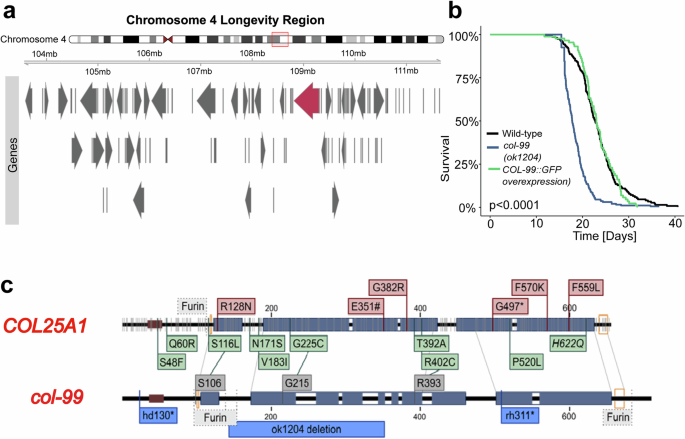
a Depiction of the human chromosome IV with the putative longevity region highlighted in red. The overlapping open reading frames located in this region are displayed in separate rows, and COL25A1 is highlighted in red (for details, see Supplementary Table 1). b Lifespan curve showing short-lived col-99(ok1204) mutant compared to wild-type. Automated Lifespan was performed using n > 87. The log-rank test was performed for statistical analysis of mean lifespans. c Schematic showing the parallels drawn between human COL25A1 and C. elegans col-99 gene and the associated SNPs. Grey lines indicate observed SNPs in COL25A1; Red boxes indicate SNPs/mutations associated with diseases, while SNPs marked in green are associated with longevity. Dark blue bars indicated collagen domains. Predicted furin sites are marked by dashed lines; orange clamps indicate cysteine-knots. SNPs in dark grey are potential longevity-associated SNPs matched on col-99. Light blue boxes are known for deletion/stop mutation in C. elegans. The Brown box indicates the transmembrane domain.
In a subsequent literature search, we found that COL25A1 exhibited several SNPs associated with exceptional lifespan and/or healthy aging (Fig. 1b, Supplementary Table 1). Most of the molecular alterations of these longevity-associated SNPs were predicted to result in the loss or reduction of the function of COL25A1. To determine whether the loss of COL25A1 is beneficial, we investigated the effect of a loss-of-function mutation in the COL25A1 orthologue col-99 in C. elegans. However, the col-99(ok1204) deletion mutant was short-lived with a mean lifespan of 18.4 days, approximately 5 days less than the wild type (Fig. 1b; blue line, Supplementary Table 2). Overexpression of full-length COL-99 protein tagged with GFP, on the other hand, did not affect the mean lifespan (Fig. 1b; green line, Supplementary Table 2). Assuming COL25A1 is responsible for the above-mentioned linkage, these findings indicate that the effect of SNPs in COL25A1 on longevity has a more complex mechanism.
S106L SNP in col-99 extends lifespan in C. elegans
To investigate this unknown mechanism, we tried to map all human SNPs in the C. elegans col-99 gene. However, due to the vast evolutionary distance and the repetitive nature of collagen, we could only reliably map three sites with some confidence (human S116L → C. elegans S106, G225C → G215, R402C → R393) (Fig. 1c). We focused on the S116L SNP in COL25A1 (S106L in col-99 in C. elegans) as it is proximal to a conserved furin cleavage site with the potential to impact domain processing (Fig. 1c).
First, to determine if the human S116L SNP introduction into col-99 in C. elegans had any effect on lifespan, we obtained C. elegans harboring this S106L SNP in col-99 (hereafter referred to as col-99(gk694263[S106L]) mutants) and outcrossed ten times (10X) to remove any potential background mutations. We then performed lifespan studies with 4X, 8X, and 10X outcrossed col-99(gk694263[S106L]) mutants and found all outcrossed col-99(gk694263[S106L]) mutants increased mean lifespans (Fig. 2a, S1b, Supplementary Table 2) by approximately 26% compared to wild-type control, indicating not only that S106L in humans might indeed be causative for prolonged life, but also re-assuring our mapping of this SNP between the species.
Fig. 2: COL-99 S106L influences longevity in C. elegans.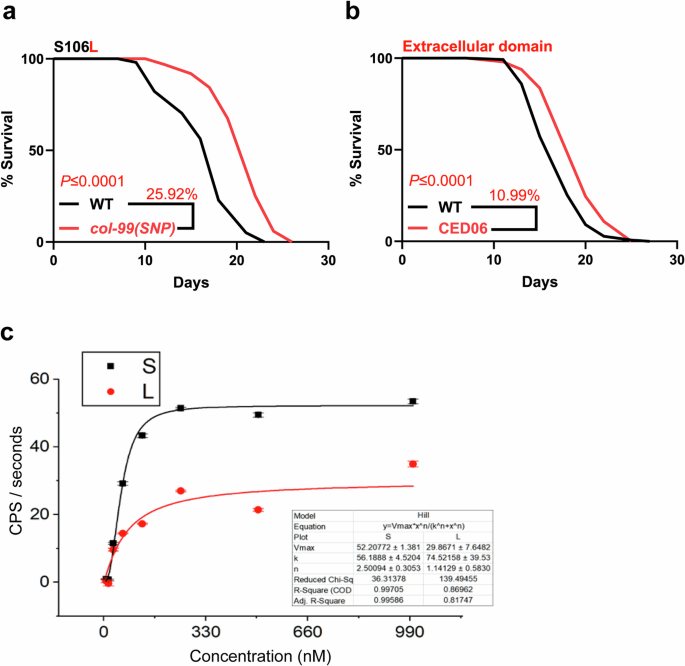
a Lifespan curve showing an extension of life span in col-99(gk694263[S106L]) SNP mutant. b Lifespan curve showing the extracellular domain (CED06 col-99(syb4332[COL-99EXT])) is sufficient to increase lifespan. The lifespans were performed in at least two biological replicates at 20 °C. Kaplan-Meier Log-Rank test was performed for statistical analysis on mean lifespans using the online software OASIS. A lifespan summary for individual experiments is provided in Supplementary Table 2. c In-vitro furin cleavage assay shows differential cleavage of COL-99 peptide. Representative data from three different assays are shown.
To independently validate that the increased lifespan is due to the single mutation in col-99, we CRISPR-generated the S106L SNP mutation into wild-type animals (hereafter referred to as col-99(syb4350[S106L])). Reassuringly, we found a similar extension in the mean lifespan of col-99(syb4350[S106L]) compared to the wild type (Fig. S1c, Supplementary Table 2). The col-99(gk694263[S106L]) mutants superficially looked wild type but were slightly delayed developmentally by a few hours, with larval L4 stage (Fig. S1d, Supplementary Table 2). Furthermore, the longevity of col-99(gk694263[S106L]) mutants did not correlate with oxidative or heat stress resilience (i.e., col-99(gk694263[S106L]) mutants were more susceptible to 14 mM arsenite or 32 °C treatment; Fig. S1e, f, Supplementary Table 2).
These results confirmed that the S106L mutation in col-99 extends lifespan independently of stress resistance pathways. As the mutation is near a conserved furin cleavage site involved in collagen processing, we next investigated whether the extracellular domain of COL-99 mediates this effect.
The extracellular domain of COL-99 is sufficient for increased longevity
COL25A1 in humans and COL-99 in C. elegans are both single-pass type II transmembrane collagen proteins with predicted furin cleavage sites7,10. For COL25A1, this site is reported to be important for shedding, creating the so-called CLAC (collagen-like Alzheimer’s amyloid plaque component) fragment11. Besides its association with Alzheimer’s Disease, the function of this shredded collagen fragment is not understood, although it seems to play a role in muscle development and innervation12,13. Similarly, in C. elegans, col-99 was important for axonal guidance, and the first and third predicted furin cleavage sites seem important for its function6,8.
As S116L (and S106L) are close to the conserved (first) furin cleavage site, we hypothesized that the S106L mutation in C. elegans might affect furin cleavage, leading to a change in the extracellular domain shedding rate. To test this hypothesis, we expressed the extracellular domain of COL-99 under its endogenous promoter in C. elegans. The expression of a shed extracellular domain of COL-99 prolonged the mean lifespan of C. elegans by approximately 11% already (Fig. 2b, Supplementary Table 2), consistent with the longevity effect of the S106L SNP in col-99(gk694263[S106L]) mutant.
Although the furin recognition site is often described as R-X-[K/R]-R⇓, the full recognition sequence is longer and amino acids up to 14 amino acids N-terminally (P14) and six amino acids C-terminally (P6′) from the cleavage site are described to influence furin activity (see Fig. 3a for clarification of P1/P1’ nomenclature and positioning in COL-99 and COL25A1)14. To measure the effect of an S to L mutation at the P2’ site in C. elegans, we synthesized short fluorescently quenched peptides containing the furin cleavage site and a serine or leucine residue, respectively. The quenching was resolved upon cleavage, and an increased fluorescence was observed (Fig. 2c). Contrary to our expectation, the mutant showed a roughly halved Vmax (30 vs 52) compared to the wild-type sequence, indicating cleavage of S106L by furin, but at a slightly decreased rate. Given the abundance of furin, this small difference can hardly explain the observed effect in vivo, especially as it seems to contradict our finding from the expression of the extracellular domain alone, which suggested benefits from increased cleavage.
Fig. 3: Phosphorylation may alter shedding of COL-99.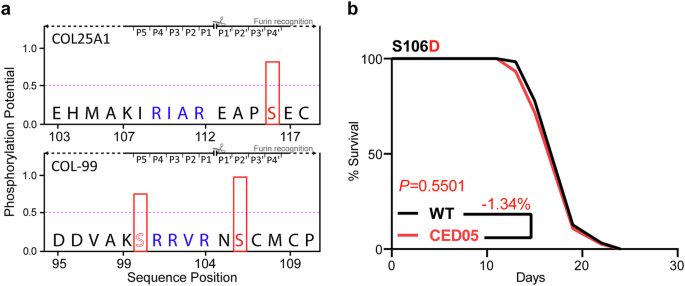
a Phosphorylation potential of serine residue in the SNP next to the furin cleavage site (highlighted in green) in COL25A1 and COL-99. Bold “Serines” are SNP S106 and S116, respectively. Prediction by NetPhos 3.1. b Lifespan curve showing incapability of phosphomimetic variant of col-99 S106D (CED05 col-99(syb4352[S106D])) to extend lifespan. The lifespan assay was performed in five independent biological replicates at 20 °C. The Kaplan-Meier Log-Rank test was performed for statistical analysis on mean lifespans using the online software OASIS. A lifespan summary for individual experiments is provided in Supplementary Table 2.
Besides its location near the furin cleavage site, both sites are also predicted to be phosphorylated (Fig. 3a). Although extracellular phosphorylation is not completely understood yet, its importance has become more evident over the last few years15,16,17,18. Bioinformatically, several phosphorylation sites in COL25A1 have been predicted19. Furthermore, it was shown at least twice that phosphorylation might affect furin cleavage20. We therefore speculated that phosphorylation of S106 might inhibit COL-99 shedding, thereby reducing the extracellular concentration of the extracellular domain of COL-99. The described SNP might prevent this phospho‑dependent inhibition, thus increasing shedding. Consequently, a CRISPR mutant of col-99 introducing a phosphomimetic S106D exchange (col-99(syb4352[S106D])) did not significantly affect the mean lifespan (3 out of 5 independent trials; Fig. 3b, Supplementary Table 2).
Taken together, this suggests a model wherein phosphorylation of serine 106 influences furin cleavage and release of the extracellular domain of COL-99 to alter downstream processes to increase lifespan.
Innate immune response is upregulated in col-99(gk694263[S106L]) mutants
To identify the downstream mechanisms affected by the S106L mutation in col-99(gk694263[S106L]), we performed transcriptomic profiling of the col-99(gk694263[S106L]) mutant and wild type at the young adult stage. The introduction of col-99 SNP drove limited transcriptomic effects (Fig. 4a, Supplementary Table 3), including a ~ 20% increase in the col-99 transcript level (Fig. 4b), suggesting an autoregulation. Gene Ontology (GO) analysis revealed a downregulation of genes in the broad “oxidation-reduction process” category (Fig. 4c, Supplementary Table 3). On closer inspection, some of these genes are core components of the mitochondrial respiratory chain and inner membrane, directly involved in ATP production. This suggests that col-99(gk694263[S106L]) mutants may have an energy production phenotype; however, we did not functionally assess mitochondrial activity, so this remains a hypothesis.
Fig. 4: Defense response genes are upregulated in col-99(gk694263[S106L]) SNP mutants.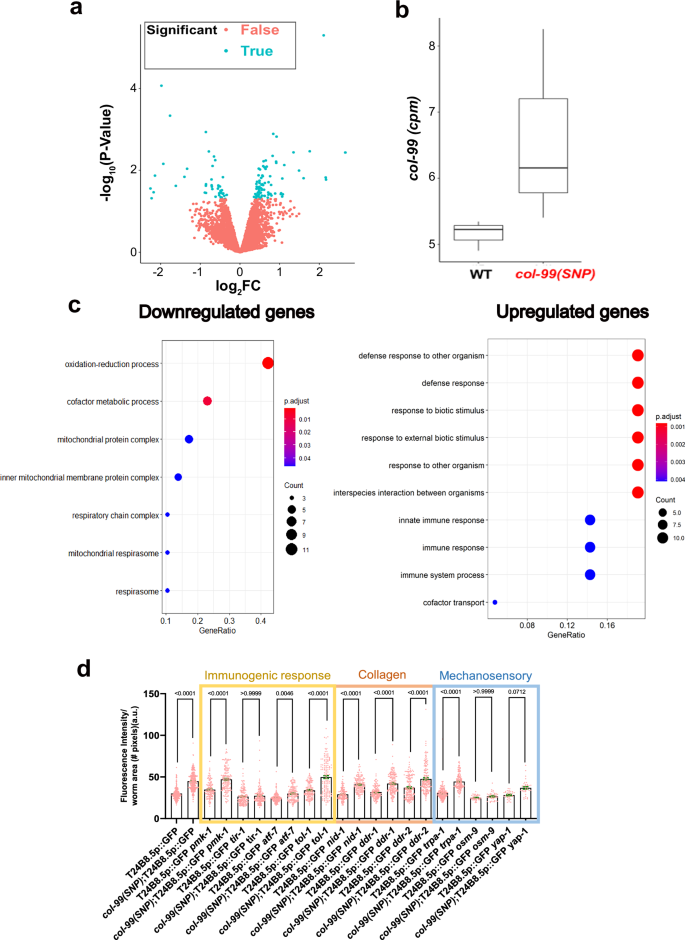
a Logarithmic scale representing the distribution of differentially expressed genes in col-99(gk694263[S106L]) SNP mutant versus wild-type. b The graph shows an increase in the expression of col-99 transcripts in col-99(gk694263[S106L]) SNP mutant compared to wild-type (in terms of FKPM values). c GO term analysis showing downregulated and upregulated genes in col-99(gk694263[S106L]) SNP mutant to wild-type. RNA sequencing was performed with three biological batches. d Targeted screening to find regulators of T24B8.5 GFP expression. Data is plotted as mean, and error bars represent SEM. Two-tailed Welch’s t-test statistical analysis. The experiment was performed in three biological batches. col-99(SNP) is col-99(gk694263[S106L]).
Meanwhile, innate immune response genes were upregulated (Fig. 4c, Supplementary Table 3). ATF-7 is the master regulator of immune response in C. elegans21, and we found it to be upregulated by col-99(gk694263[S106L]) expression (Fig. S2a, Supplementary Table 3). However, in our epistatic lifespan analysis, we observed that atf-7 was not required for mean lifespan extension in col-99(gk694263[S106L]) mutants (Fig. S2b, Supplementary Table 2). Arginine kinase (creatine kinase) argk-1 was also upregulated in col-99(gk694263[S106L]) mutants (Supplementary Table 3). We verified this increase in col-99(gk694263[S106L]) mutants with a reporter strain argk-1p::GFP using fluorescence microscopy (Fig. S2c). Previously, ARGK-1 overexpression was shown to increase lifespan via AMPK, which triggers the activation of the innate immune response22,23. Therefore, we checked for the requirement of argk-1 in lifespan extension by col-99(gk694263[S106L]) mutants. We assayed the lifespan of argk-1(ok2993); col-99(gk694263[S106L]) double mutants but found that the extended lifespan of col-99(gk694263[S106L]) mutant was independent of argk-1 (Fig. S2d, Supplementary Table 2). Since DAF-16 is also an important modulator of innate immune response and lifespan24, we tested the nuclear localization of DAF-16 in col-99(gk694263[S106L]) mutants. However, the differences in DAF-16 nuclear localization were insignificant (Fig. S2e). Taken together, our approach in testing these genes, differentially expressed in RNA sequencing, did not identify underlying pathways driving longevity.
Next, we employed a candidate RNAi screen approach to identify the pathways mediating the lifespan extension of col-99(gk694263[S106L]). Given the strong innate immune transcriptional signature, we tested two immune-response reporter genes, clec-85 and T24B8.5, that were also differentially regulated by col-99(gk694263[S106L]) (Fig. 4d, S2f, Supplementary Table 3). The transcriptional reporter driving GFP under the T24B8.5 promoter (T24B8.5p::GFP25) showed a two-fold upregulation in the fluorescence intensity in col-99(gk694263[S106L]) mutants compared to wild-type, making it suitable for screening. Given that col-99(gk694263[S106L]) induces T24B8.5p::GFP in vivo, knocking down genes that are required for mediating col-99(gk694263[S106L])-dependent T24B8.5 upregulation may reveal underlying pathways. Therefore, we screened through a targeted candidate library including known immunogenic-, collagen-, and mechano-receptors using the T24B8.5p::GFP reporter strain.
We started with genes in the canonical innate immune response pathway (P38 MAP Kinase pmk-126, Toll and Interleukin 1 Receptor tir-127, ATF cAMP-dependent transcription factor atf-728, and Toll-like receptor tol-129) and found that RNAi targeting tir-1 suppressed the col-99(gk694263[S106L])-mediated T24B8.5p::GFP induction (Fig. 4d), indicating that this immune receptor was required. However, it is unlikely that the extracellular domain of COL-99 would bind the TIR-1 receptor directly for signaling, suggesting additional upstream signaling. To identify the potential direct binding partners/receptors of shed COL-99 extracellular domain, we turned to the COL-99 collagen receptors, the Discoidin Domain Receptors (ddr-1 and ddr-2), and basement membrane component nidogen homolog nid-1, which are putative binding partners of the cleaved COL-99 ectodomain and are involved in col-99-mediated neuronal axon guidance6. However, knockdown of ddr-1, ddr-2, or nid-1 did not suppress col-99(gk694263[S106L])-mediated T24B8.5p::GFP induction (Fig. 4d). COL-99 is expressed in the hypodermis, and the furin cleavage and shedding of the COL-99 ectodomain are required for neuronal axon guidance6. Thus, a neuronal receptor may be required. We tested neuronal and mechanosensitive transient receptor potential (TRP) channel trpa-1/TRPA and osm-9/TRPV, which are shown to be involved in longevity30,31. We found that osm-9 was required for col-99(gk694263[S106L])-mediated T24B8.5p::GFP induction (Fig. 4d). OSM-9 is a mechanosensitive receptor upstream of YAP-1 (yes-associated protein), a mechanoresponsive transcriptional co-activator activated upon stiffness changes in the extracellular collagen network and implicated in the control of lifespan32,33,34. In addition, yap-1 is functionally implicated in Touch Receptor Neuron asymmetric neurite extension35 and pathogen resistance in C. elegans36, consistent with a potential role for neuronal regulation and innate immunity. We found that the knockdown of yap-1 also suppressed col-99(gk694263[S106L])-mediated T24B8.5p::GFP induction (Fig. 4d). Taken together, we identified two receptors, tir-1 and osm-9, and the co-transcriptional activator yap-1 required for col-99(gk694263[S106L]) induction of the transcriptional innate immune response program.
Since SNP could possibly affect the folding or trimerization of COL-99 collagen, we also included the hsp-4::GFP reporter as part of a broader mechanistic screen for stress-responsive pathways, to assess whether col-99 SNP might affect the unfolded protein response (UPR), despite no prior evidence linking COL-99 to ER stress. By contrast to the immune-related reporter screen, yap-1 and tir-1 were not required for col-99(gk694263[S106L]) induction of hsp-4 (Fig. S2g, h), suggesting that our reporter screen findings need to be validated with functional lifespan assays.
The P38 MAPK pathway is required for longevity extension by col-99(gk694263[S106L])
Given the distinct downstream pathways we recovered for col-99(gk694263[S106L]), we aimed to determine the pathway that mediates col-99(gk694263[S106L]) longevity. For instance, our reporter assays revealed that the COL-99 receptors ddr-1 and ddr-2 were not required for immune and UPR responses; this does not rule out their role in lifespan regulation. However, we found that loss of ddr-1, or ddr-2, or the double mutant ddr-1; ddr-2 did not suppress col-99(gk694263[S106L])-induced longevity (Fig. S3a, Supplementary Table 2), suggesting that DDR-1 and DDR-2 are the COL-99 receptor for axonal guidance6 but not for longevity. Another likely candidate for COL-99 ectodomain receptor is TIR-1, and we found that tir-1 was required for col-99(gk694263[S106L])-induced lifespan extension (Fig. 5a, Supplementary Table 2). Since tir-1 is upstream of the p38 MAPK signaling cascade, we tested whether the downstream protein p38 MAPK (pmk-1) was required for lifespan extension. Although RNAi of pmk-1 was not sufficient to block T24B8.5p::GFP induction in col-99(gk694263[S106L]) mutants, knockdown of pmk-1 abolished the longevity of col-99(gk694263[S106L]) mutants (Fig. 5b, Supplementary Table 2). We confirmed these lifespan results of col-99(gk694263[S106L]) with the CRISPR-generated mutant CED04 col-99(syb4350[S106L]), and also showed no lifespan extension with the phosphomimetic CED05 col-99(syb4352[S106D]) mutants in combination with tir-1 or pmk-1 knockdown (Fig. S3b–e, Supplementary Table 2). Consistently, tir-1 and pmk-1 were required for lifespan extension in the CED06 col-99(syb4332[COL-99EXT]) mutant expressing the ectodomain of COL-99 (Fig. 5c, d, Supplementary Table 2).
Fig. 5: p38 MAPK is responsible for extending lifespan in col-99(gk694263[S106L]) SNP mutants.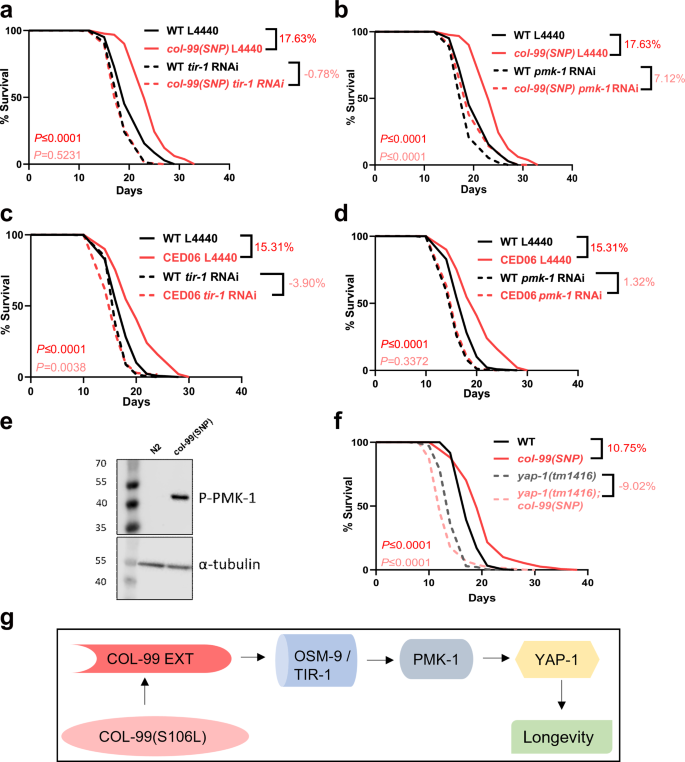
Survival curve showing the dependence of the increased lifespan of col-99(gk694263[S106L]) SNP mutant on tir-1(a), pmk-1(b). Survival curve showing the dependence of the increased lifespan of CED06 col-99(syb4350[S106L]) SNP mutant on tir-1(c) and pmk-1 (d). e Western Blot showing activation of PMK-1 in col-99(gk694263[S106L]) SNP mutant. f Survival curve showing the dependence of col-99(gk694263[S106L]) SNP on yap-1 for extension of lifespan. g Hypothetical model of signal relay from the extracellular domain of COL-99 to activate the p38 MAPK pathway, which signals to YAP-1 to extend longevity in the col-99(gk694263[S106L]) SNP mutant. Genes displayed are unlikely to be in the same cell. Please, also see Fig. S4. The lifespans were performed in at least two biological replicates at 20 °C. Kaplan-Meier Log-Rank test was performed for statistical analysis on mean lifespans using the online software OASIS. The Western blot was done twice. A lifespan summary and raw western blots for individual experiments are provided in Supplementary Table 2. col-99(SNP) is col-99(gk694263[S106L]).
Furthermore, in line with the requirement of pmk-1 for lifespan extension, col-99(gk694263[S106L]) mutants showed higher p38 MAPK/PMK-1 phosphorylation (Fig. 5e, Supplementary Table 2), an event generally known to drive increased ATF-7 transcription factor levels. Despite this association, ATF-7 was not required for the extended lifespan in col-99(gk694263[S106L]) mutants (Fig. S2b, Supplementary Table 2). This led us to investigate other potential mediators, and we discovered that YAP-1 played a critical role in facilitating the longevity effects of col-99(gk694263[S106L]) mutants (Fig. 5f, Supplementary Table 2). Collectively, our data support a model in which a non-canonical immune response pathway involving osm-9, tir-1, pmk-1, and yap-1 is activated upon blunting the phosphorylation of COL-99 at the S106 site, and thus altered furin-mediated cleavage resulting in the subsequent release of the COL-99 ectodomain (Fig.5g, S4).